Creating a topology file
Overview
Teaching: 30 min
Exercises: 0 minQuestions
How do I create a topology file for my system?
Objectives
Understand packmol input files
Use VMD to generate a LAMMPS data file
PACKMOL
PACKMOL can create an initial point for MD simulations by packing molecules in defined regions of space. Installation instructions can be found in the user guide.
To pack a new system you will need a packmol input script and topology files for however many molecules your system has.
The molecule topology files are commonly .xyz files, but packmol also accepts .pdb, moldy, and tinker.
As an example in this lesson, we will use water.xyz and ethanol.xyz, see the end of this section for the full file.
PACKMOL input script
A minimum input script must contain:
The distance tolerance required
tolerance 2.0
measured in angstrom, 2.0 is a good value for atomistic simulations, you may want to use higher values for coarse-grained systems.
The name of and file type for the output file:
output water_ethanol_mix.xyz
filetype xyz
the default file type is pdb, packmol also accepts xyz, tinker, and moldy.
And at least one molecule, using a structure ... end structure section.
In this example, we will have two.
structure water.xyz
number 1300
inside cube 15. 15. 30. 50.
end structure
The first line starts the structure section, and selects which file to read the molecule from, in this case water.xyz.
Then number selects how many molecules the final system will have.
The next line, the constraint, selects how the molecules will be arranged in the final system, in this case, on a cube, with side 50 angstrom, and with origin at (15, 15, 30).
You can then run the script using the packmol command as so:
packmol < pack.inp
This results in a cube of water molecules as so:

There are several types of constraints available: fixed, constrain rotations, combinations of inside/outside with cube/box/sphere/ellipsoid/cylinder, as well as combinations of above/below with plane/xgauss.
Descriptions of all of these types can be found in the manual.
You can also combine constrains, for example, here we constrain the ethanol molecules to be in a square prism, with 1 < x < 79, 1 < y < 79, and 1 < z < 109, but outside the cube where the water molecules are.
structure ethanol.xyz
number 1300
inside box 1. 1. 1. 79. 79. 109.
outside cube 15. 15. 30. 50.
end structure
The extra angstrom around the outermost limits allows the later application of periodic boundary conditions without creating bonds between atoms of different molecules.
The resulting system looks like the following:
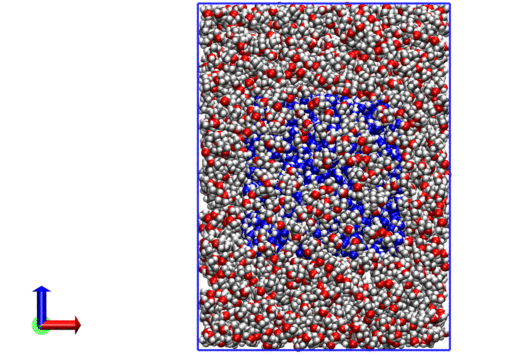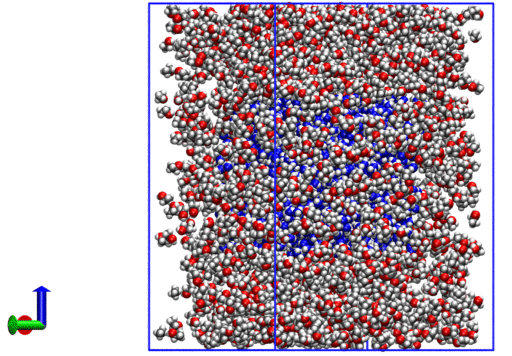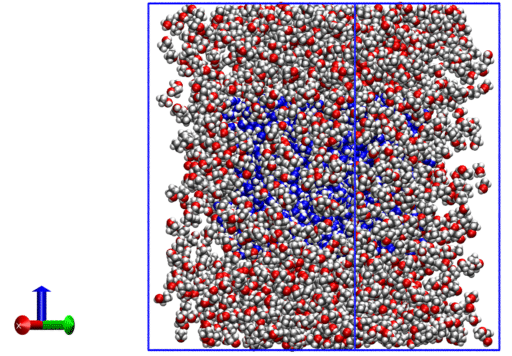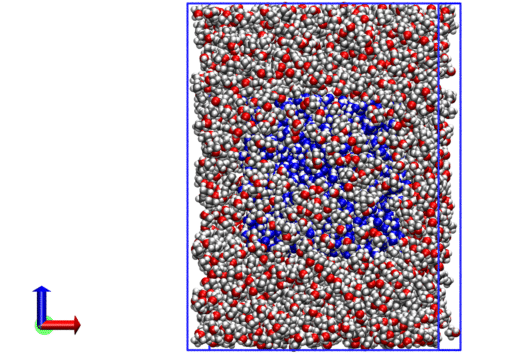
We now have a .xyz with coordinates for all molecules.
In the next section, we will use VMD to automatically create the bonds, angles, and dihedral connections in each molecule, and write a LAMMPS data file.
Tip
To solvate large molecules/particles (for example, polymers), it is considerably faster to do multi-step packing, with different input files, one per molecule type, and using the
fixedconstraint for the topology with the already packed molecules.Example:
tolerance 2.0 output polymers_with_water.xyz filetype xyz structure packed_polymers.xyz number 1 fixed 0. 0. 0. 0. 0. 0. end structure structure water.xyz number 1300 inside cube 1. 1. 1. 99. end structure
water.xyz
The topology file for water.
Expand for full file
3 Water molecule WO 0.00000 0.00000 0.11779 WH 0.00000 0.75545 -0.47116 WH 0.00000 -0.75545 -0.47116
ethanol.xyz
The topology file for ethanol.
Expand for full file
9 Ethanol EHC 1.8853 -0.0401 1.0854 ECH 1.2699 -0.0477 0.1772 EHC 1.5840 0.8007 -0.4449 EHC 1.5089 -0.9636 -0.3791 ECO -0.2033 0.0282 0.5345 EHA -0.4993 -0.8287 1.1714 EHA -0.4235 0.9513 1.1064 EOH -0.9394 0.0157 -0.6674 EHO -1.8540 0.0626 -0.4252
pack.inp
The PACKMOL input script
Expand for full file
tolerance 2.0 output water_ethanol.xyz filetype xyz structure water.xyz number 1300 inside cube 15. 15. 30. 50. end structure structure ethanol.xyz number 1300 inside box 1. 1. 1. 79. 79. 109. outside cube 15. 15. 30. 50. end structure
Converting a .xyz file to a LAMMPS data file with VMD
To do this, you will need access to working VMD binary.
Almost every command shown in this section can be used directly in the tk console (Extensions > TK Console), or in the terminal where VMD was called from.
However, here we will show how to write them in text files, and then having VMD sourcing (reading) the files and running all the commands in succession.
The overall process to follow is:
- For each atom type:
- Select all atoms from a given atomtype.
- Assign relevant properties to all of the selected atoms (mass, type, charge, element, radius).
- When all properties are assigned:
- Guess bonds, angles, dihedrals.
- Confirm the reported number of connections (bonds, angles, dihedrals) and types are correct.
- Write the result as a LAMMPS data file.
Since we have two types of molecules in our system, there are two files with commands to assign properties to each molecule’s atoms (ethanol.tcl and water.tcl), and one file with the commands to generate the bond, angle, and dihedral information, as other auxiliary information.
Tip
You can have all of the commands in one single file, but keeping each molecule in a separate file allows for simpler re-use of
tclscripts in later system creation.
The first thing to do is to load the water_ethanol.xyz file we created in the previous step.
This can be done in various ways, the easiest is to call VMD on the correct file in the terminal with vmd water_ethanol.xyz.
The sections to select and assign properties for each atom types are all similar, only with different values for each property, so we will only analyse one example, WO from the water.tcl file:
set selwo [atomselect top {name WO}]
$selwo set mass 15.999
$selwo set type WO
$selwo set charge -0.8340
$selwo set element O
$selwo set radius 1.2
The first line can divided into its two constituent commands set selwo <selection> and atomselect top {name WO}.
Let us analyse them in reverse order.
The atomselect command returns the name of a selection of atoms, and can accept a range of selection methods.
In the example above, we are selecting all atoms from the top file (the currently selected file in VMD, the only one in this case) named WO (the first column in .xyz files are used as names in VMD).
The set selwo <selection> command takes the named selection and assigns it to a variable named selwo.
The following lines are all very similar, they all assign a certain value to each property to every atom in the selwo selection – mass, type, charge, element, or radius.
The correct masses and charges will be necessary to create a LAMMPS data file, the radius will be used to generate bonds/angles/dihedrals in the next step, and the element/type are useful in visual atom selections to check if your generated bonds/angles/dihedrals are correct.
To actually apply these commands to our system, we use the source command in either the terminal where VMD was called, or in the TK Console:
source water.tcl
source ethanol.tcl
With every atomic property set, we can then use VMD’s topotools to generate bond/angle/dihedral information, using the commands in topo.tcl, which we shall now analyse line by line.
If you launch VMD’s TK Console, it will load topotools by default, but if you use the source command, you need to tell the tcl shell to load it, with:
package require topotools
Then, there is a definition for a function that calculates the total charge of the system – wrong charge in a LAMMPS simulation is a very common source of crashes and/or bad simulation results.
proc get_total_charge {
eval "vecadd [[atomselect $molid all] get charge]"
}
Now, we use the next two commands to recalculate the bonds, using the radii set in the two .tcl files we sources previously, and to re-name them using the scheme atom1name-atom2name
mol bondsrecalc all
topo retypebonds
And then we use the topotools package to generate angle and dihedral information, from the already formed bonds:
topo guessangles
topo guessdihedrals
The remainder of the lines just output useful information to the command line, like the number of bonds and bondtypes, things that you should calculate by yourself beforehand and confirm that matches with the VMD output.
It also outputs the calculated total charge for the system - this should be close to zero for neutral systems, but due to floating point errors it will probably not be exactly zero, but something like -6.705522537231445e-5.
The information about bond/angle/dihedral typenames can be useful to identify where wrong bonds are being created, and you might then need to adjust the size of an atom type radius, or re-pack your molecules on a larger box.
Finally, the last line writes a LAMMPS data file.
topo writelammpsdata data.water_ethanol
Tip
If you have already validated your data file generation, or you know how to judge whether the number of bond/angle/dihedrals and their types are the correct amount, you can launch VMD without a graphical interface with the
-dispdevflag. This can be useful if you’re packing a large system on a remote computer.vmd -dispdev text water_ethanol.xyz
Important note
The data file generated with these steps is still missing the force field parameters that LAMMPS needs to simulate a system. You will need to define these either on the data file you just created (un-comment and fill in the Pair/Bond/Angle/Dihedral coeffs sections) or in the LAMMPS input file, like we did in earlier lessons.
ethanol.tcl
The
tclfile for the ethanol molecule.Expand for full file
set selECH [atomselect top {name ECH}] $selECH set mass 12.011 $selECH set type ECH $selECH set charge -0.18 $selECH set element C $selECH set radius 1.4 set selECO [atomselect top {name ECO}] $selECO set mass 12.011 $selECO set type ECO $selECO set charge 0.145 $selECO set element C $selECO set radius 1.4 set selEHC [atomselect top {name EHC}] $selEHC set mass 1.008 $selEHC set type EHC $selEHC set charge 0.06 $selEHC set element H $selEHC set radius 0.8 set selEHA [atomselect top {name EHA}] $selEHA set mass 1.008 $selEHA set type EHA $selEHA set charge 0.06 $selEHA set element H $selEHA set radius 0.8 set selEHO [atomselect top {name EHO}] $selEHO set mass 1.008 $selEHO set type EHO $selEHO set charge 0.418 $selEHO set element H $selEHO set radius 0.8 set selEOH [atomselect top {name EOH}] $selEOH set mass 15.999 $selEOH set type EOH $selEOH set charge -0.683 $selEOH set element O $selEOH set radius 1.4
water.tcl
The
tclfile for the water molecule.Expand for full file
set selwo [atomselect top {name WO}] $selwo set mass 15.999 $selwo set type WO $selwo set charge -0.8340 $selwo set element O $selwo set radius 1.2 set selwh [atomselect top {name WH}] $selwh set mass 1.008 $selwh set type WH $selwh set charge 0.4170 $selwh set element H $selwh set radius 0.8
topo.tcl
The
tclfile that generates the bond/angle/dihedral information and writes the LAMMPS datafile.Expand for full file
package require topotools proc get_total_charge { eval "vecadd [[atomselect $molid all] get charge]" } mol bondsrecalc all topo retypebonds topo guessangles topo guessdihedrals puts "" puts "Bond type names: " puts [ topo bondtypenames ] puts "" puts "Angle type names: " puts [ topo angletypenames ] puts "" puts "Dihedral type names: " puts [ topo dihedraltypenames ] puts "" puts "" puts [format "Number of bonds: %s" [topo numbonds] ] puts [format "Number of bonds types: %s" [topo numbondtypes] ] puts [format "Number of angles: %s" [topo numangles] ] puts [format "Number of angles types: %s" [topo numangletypes] ] puts [format "Number of dihedral: %s" [topo numdihedrals] ] puts [format "Number of dihedral types: %s" [topo numdihedraltypes] ] puts [format "Total charge: %s" [get_total_charge] ] puts "" topo writelammpsdata data.water_ethanol
Key Points
PACKMOL allows the generation of randomly packed systems from single-molecule XYZ files
VMD’s topotools can generate bond, angle, and dihedral information from user-defined properties