Content from Why use an HPC System?
Last updated on 2025-01-14 | Edit this page
Estimated time: 20 minutes
Overview
Questions
- Why would I be interested in High Performance Computing (HPC)?
- What can I expect to learn from this course?
Objectives
- Be able to describe what an HPC system is
- Identify how an HPC system could benefit you.
HPC research examples
Frequently, research problems that use computing can outgrow the capabilities of the desktop or laptop computer where they started:
- A statistics student wants to cross-validate a model. This involves running the model 1000 times — but each run takes an hour. Running the model on a laptop will take over a month! In this research problem, final results are calculated after all 1000 models have run, but typically only one model is run at a time (in serial) on the laptop. Since each of the 1000 runs is independent of all others, and given enough computers, it’s theoretically possible to run them all at once (in parallel).
- A genomics researcher has been using small datasets of sequence data, but soon will be receiving a new type of sequencing data that is 10 times as large. It’s already challenging to open the datasets on a computer — analyzing these larger datasets will probably crash it. In this research problem, the calculations required might be impossible to parallelize, but a computer with more memory would be required to analyze the much larger future data set.
- An engineer is using a fluid dynamics package that has an option to run in parallel. So far, this option was not utilized on a desktop. In going from 2D to 3D simulations, the simulation time has more than tripled. It might be useful to take advantage of that option or feature. In this research problem, the calculations in each region of the simulation are largely independent of calculations in other regions of the simulation. It’s possible to run each region’s calculations simultaneously (in parallel), communicate selected results to adjacent regions as needed, and repeat the calculations to converge on a final set of results. In moving from a 2D to a 3D model, both the amount of data and the amount of calculations increases greatly, and it’s theoretically possible to distribute the calculations across multiple computers communicating over a shared network.
In all these cases, access to more (and larger) computers is needed. Those computers should be usable at the same time, solving many researchers’ problems in parallel.
Break the Ice
Talk to your neighbour, office mate or rubber duck about your research.
- How does computing help you do your research?
- How could more computing help you do more or better research?
A Standard Laptop for Standard Tasks
Today, people coding or analysing data typically work with laptops.

Let’s dissect what resources programs running on a laptop require:
- the keyboard and/or touchpad is used to tell the computer what to do (Input)
- the internal computing resources Central Processing Unit and Memory perform calculation
- the display depicts progress and results (Output)
Schematically, this can be reduced to the following:

When Tasks Take Too Long
When the task to solve becomes heavy on computations, the operations are typically out-sourced from the local laptop or desktop to elsewhere. Take for example the task to find the directions for your next vacation. The capabilities of your laptop are typically not enough to calculate that route spontaneously: finding the shortest path through a network runs on the order of (v log v) time, where v (vertices) represents the number of intersections in your map. Instead of doing this yourself, you use a website, which in turn runs on a server, that is almost definitely not in the same room as you are.

Note here, that a server is mostly a noisy computer mounted into a rack cabinet which in turn resides in a data center. The internet made it possible that these data centers do not require to be nearby your laptop. What people call the cloud is mostly a web-service where you can rent such servers by providing your credit card details and requesting remote resources that satisfy your requirements. This is often handled through an online, browser-based interface listing the various machines available and their capacities in terms of processing power, memory, and storage.
The server itself has no direct display or input methods attached to it. But most importantly, it has much more storage, memory and compute capacity than your laptop will ever have. In any case, you need a local device (laptop, workstation, mobile phone or tablet) to interact with this remote machine, which people typically call ‘a server’.
When One Server Is Not Enough
If the computational task or analysis to complete is daunting for a single server, larger agglomerations of servers are used. These go by the name of “clusters” or “super computers”.

The methodology of providing the input data, configuring the program options, and retrieving the results is quite different to using a plain laptop. Moreover, using a graphical interface is often discarded in favor of using the command line. This imposes a double paradigm shift for prospective users asked to
- work with the command line interface (CLI), rather than a graphical user interface (GUI)
- work with a distributed set of computers (called nodes) rather than the machine attached to their keyboard & mouse
I’ve Never Used a Server, Have I?
Take a minute and think about which of your daily interactions with a computer may require a remote server or even cluster to provide you with results.
- Checking email: your computer (possibly in your pocket) contacts a remote machine, authenticates, and downloads a list of new messages; it also uploads changes to message status, such as whether you read, marked as junk, or deleted the message. Since yours is not the only account, the mail server is probably one of many in a data center.
- Searching for a phrase online involves comparing your search term against a massive database of all known sites, looking for matches. This “query” operation can be straightforward, but building that database is a monumental task! Servers are involved at every step.
- Searching for directions on a mapping website involves connecting
your
- starting and (B) end points by traversing a graph in search of the “shortest” path by distance, time, expense, or another metric. Converting a map into the right form is relatively simple, but calculating all the possible routes between A and B is expensive.
Checking email could be serial: your machine connects to one server and exchanges data. Searching by querying the database for your search term (or endpoints) could also be serial, in that one machine receives your query and returns the result. However, assembling and storing the full database is far beyond the capability of any one machine. Therefore, these functions are served in parallel by a large, “hyperscale” collection of servers working together.
Key Points
- “High Performance Computing (HPC) typically involves connecting to very large computing systems elsewhere in the world.”
- “These other systems can be used to do work that would either be impossible or much slower on smaller systems.”
- “The standard method of interacting with such systems is via a command line interface called Bash.”
Content from Working on a remote HPC system
Last updated on 2025-01-14 | Edit this page
Estimated time: 35 minutes
Overview
Questions
- What is an HPC system?
- How does an HPC system work?
- How do I log on to a remote HPC system?
Objectives
- Connect to a remote HPC system.
- Understand the general HPC system architecture.
What Is an HPC System?
The words “cloud”, “cluster”, and the phrase “high-performance computing” or “HPC” are used a lot in different contexts and with various related meanings. So what do they mean? And more importantly, how do we use them in our work?
The cloud is a generic term commonly used to refer to computing resources that are a) provisioned to users on demand or as needed and b) represent real or virtual resources that may be located anywhere on Earth. For example, a large company with computing resources in Brazil, Zimbabwe and Japan may manage those resources as its own internal cloud and that same company may also utilize commercial cloud resources provided by Amazon or Google. Cloud resources may refer to machines performing relatively simple tasks such as serving websites, providing shared storage, providing web services (such as e-mail or social media platforms), as well as more traditional compute intensive tasks such as running a simulation.
The term HPC system, on the other hand, describes a stand-alone resource for computationally intensive workloads. They are typically comprised of a multitude of integrated processing and storage elements, designed to handle high volumes of data and/or large numbers of floating-point operations (FLOPS) with the highest possible performance. For example, all of the machines on the Top-500 list are HPC systems. To support these constraints, an HPC resource must exist in a specific, fixed location: networking cables can only stretch so far, and electrical and optical signals can travel only so fast.
The word “cluster” is often used for small to moderate scale HPC resources less impressive than the Top-500. Clusters are often maintained in computing centers that support several such systems, all sharing common networking and storage to support common compute intensive tasks.
Logging In
The first step in using a cluster is to establish a connection from our laptop to the cluster. When we are sitting at a computer (or standing, or holding it in our hands or on our wrists), we have come to expect a visual display with icons, widgets, and perhaps some windows or applications: a graphical user interface, or GUI. Since computer clusters are remote resources that we connect to over often slow or laggy interfaces (WiFi and VPNs especially), it is more practical to use a command-line interface, or CLI, in which commands and results are transmitted via text, only. Anything other than text (images, for example) must be written to disk and opened with a separate program.
If you have ever opened the Windows Command Prompt or macOS Terminal, you have seen a CLI. If you have already taken The Carpentries’ courses on the UNIX Shell or Version Control, you have used the CLI on your local machine somewhat extensively. The only leap to be made here is to open a CLI on a remote machine, while taking some precautions so that other folks on the network can’t see (or change) the commands you’re running or the results the remote machine sends back. We will use the Secure SHell protocol (or SSH) to open an encrypted network connection between two machines, allowing you to send & receive text and data without having to worry about prying eyes.

Make sure you have a SSH client installed on your laptop. Refer to
the setup section for more details. SSH clients
are usually command-line tools, where you provide the remote machine
address as the only required argument. If your username on the remote
system differs from what you use locally, you must provide that as well.
If your SSH client has a graphical front-end, such as PuTTY or
MobaXterm, you will set these arguments before clicking “connect.” From
the terminal, you’ll write something like
ssh userName@hostname, where the “@” symbol is used to
separate the two parts of a single argument.
Go ahead and open your terminal or graphical SSH client, then log in to the cluster using your username and the remote computer you can reach from the outside world, EPCC, The University of Edinburgh.
Remember to replace userid with your username or the one
supplied by the instructors. You may be asked for your password. Watch
out: the characters you type after the password prompt are not displayed
on the screen. Normal output will resume once you press
Enter.
Where Are We?
Very often, many users are tempted to think of a high-performance
computing installation as one giant, magical machine. Sometimes, people
will assume that the computer they’ve logged onto is the entire
computing cluster. So what’s really happening? What computer have we
logged on to? The name of the current computer we are logged onto can be
checked with the hostname command. (You may also notice
that the current hostname is also part of our prompt!)
What’s in Your Home Directory?
The system administrators may have configured your home directory
with some helpful files, folders, and links (shortcuts) to space
reserved for you on other filesystems. Take a look around and see what
you can find. Hint: The shell commands pwd and
ls may come in handy. Home directory contents vary from
user to user. Please discuss any differences you spot with your
neighbors.
The deepest layer should differ: userid is uniquely
yours. Are there differences in the path at higher levels?
If both of you have empty directories, they will look identical. If you or your neighbor has used the system before, there may be differences. What are you working on?
Use pwd to print the
working directory path:
You can run ls to list
the directory contents, though it’s possible nothing will show up (if no
files have been provided). To be sure, use the -a flag to
show hidden files, too.
At a minimum, this will show the current directory as .,
and the parent directory as ...
Nodes
Individual computers that compose a cluster are typically called nodes (although you will also hear people call them servers, computers and machines). On a cluster, there are different types of nodes for different types of tasks. The node where you are right now is called the head node, login node, landing pad, or submit node. A login node serves as an access point to the cluster.
As a gateway, it is well suited for uploading and downloading files, setting up software, and running quick tests. Generally speaking, the login node should not be used for time-consuming or resource-intensive tasks. You should be alert to this, and check with your site’s operators or documentation for details of what is and isn’t allowed. In these lessons, we will avoid running jobs on the head node.
Dedicated Transfer Nodes
If you want to transfer larger amounts of data to or from the cluster, some systems offer dedicated nodes for data transfers only. The motivation for this lies in the fact that larger data transfers should not obstruct operation of the login node for anybody else. Check with your cluster’s documentation or its support team if such a transfer node is available. As a rule of thumb, consider all transfers of a volume larger than 500 MB to 1 GB as large. But these numbers change, e.g., depending on the network connection of yourself and of your cluster or other factors.
The real work on a cluster gets done by the worker (or compute) nodes. Worker nodes come in many shapes and sizes, but generally are dedicated to long or hard tasks that require a lot of computational resources.
All interaction with the worker nodes is handled by a specialized piece of software called a scheduler (the scheduler used in this lesson is called Slurm). We’ll learn more about how to use the scheduler to submit jobs next, but for now, it can also tell us more information about the worker nodes.
For example, we can view all of the worker nodes by running the
command sinfo.
OUTPUT
PARTITION AVAIL TIMELIMIT NODES STATE NODELIST
standard up 1-00:00:00 27 drain* nid[001029,001050,001149,001363,001366,001391,001552,001568,001620,001642,001669,001672-001675,001688,001690-001691,001747,001751,001783,001793,001812,001832-001835]
standard up 1-00:00:00 5 down* nid[001024,001026,001064,001239,001898]
standard up 1-00:00:00 8 drain nid[001002,001028,001030-001031,001360-001362,001745]
standard up 1-00:00:00 945 alloc nid[001000-001001,001003-001023,001025,001027,001032-001037,001040-001049,001051-001063,001065-001108,001110-001145,001147,001150-001238,001240-001264,001266-001271,001274-001334,001337-001359,001364-001365,001367-001390,001392-001551,001553-001567,001569-001619,001621-001637,001639-001641,001643-001668,001670-001671,001676,001679-001687,001692-001734,001736-001744,001746,001748-001750,001752-001782,001784-001792,001794-001811,001813-001824,001826-001831,001836-001890,001892-001897,001899-001918,001920,001923-001934,001936-001945,001947-001965,001967-001981,001984-001991,002006-002023]
standard up 1-00:00:00 37 resv nid[001038-001039,001109,001146,001148,001265,001272-001273,001335-001336,001638,001677-001678,001735,001891,001919,001921-001922,001935,001946,001966,001982-001983,001992-002005] There are also specialized machines used for managing disk storage, user authentication, and other infrastructure-related tasks. Although we do not typically logon to or interact with these machines directly, they enable a number of key features like ensuring our user account and files are available throughout the HPC system.
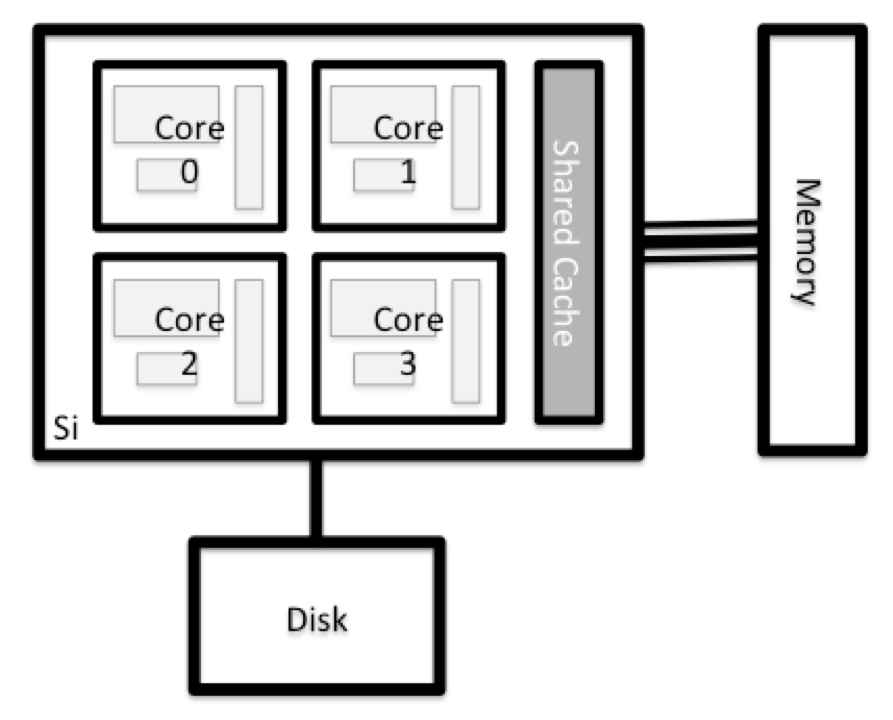
What's in a Node?
All of the nodes in an HPC system have the same components as your own laptop or desktop: CPUs (sometimes also called processors or cores), memory (or RAM), and disk space. CPUs are a computer’s tool for actually running programs and calculations. Information about a current task is stored in the computer’s memory. Disk refers to all storage that can be accessed like a file system. This is generally storage that can hold data permanently, i.e. data is still there even if the computer has been restarted. While this storage can be local (a hard drive installed inside of it), it is more common for nodes to connect to a shared, remote fileserver or cluster of servers.
There are several ways to do this. Most operating systems have a graphical system monitor, like the Windows Task Manager. More detailed information can sometimes be found on the command line. For example, some of the commands used on a Linux system are:
Run system utilities
Read from /proc
Run system monitor
Explore the login node
Now compare the resources of your computer with those of the head node.
BASH
[user@laptop ~]$ ssh userid@login.archer2.ac.uk
userid@ln03:~> nproc --all
userid@ln03:~> free -mYou can get more information about the processors using
lscpu, and a lot of detail about the memory by reading the
file /proc/meminfo:
You can also explore the available filesystems using df
to show disk free space. The
-h flag renders the sizes in a human-friendly format, i.e.,
GB instead of B. The type flag -T shows
what kind of filesystem each resource is.
Discussion
The local filesystems (ext, tmp, xfs, zfs) will depend on whether you’re on the same login node (or compute node, later on). Networked filesystems (beegfs, cifs, gpfs, nfs, pvfs) will be similar — but may include userid, depending on how it is mounted.
Compare Your Computer, the login node and the compute node
Compare your laptop’s number of processors and memory with the numbers you see on the cluster head node and worker node. Discuss the differences with your neighbor.
What implications do you think the differences might have on running your research work on the different systems and nodes?
Differences Between Nodes
Many HPC clusters have a variety of nodes optimized for particular workloads. Some nodes may have larger amount of memory, or specialized resources such as Graphical Processing Units (GPUs).
With all of this in mind, we will now cover how to talk to the cluster’s scheduler, and use it to start running our scripts and programs!
Key Points
- “An HPC system is a set of networked machines.”
- “HPC systems typically provide login nodes and a set of worker nodes.”
- “The resources found on independent (worker) nodes can vary in volume and type (amount of RAM, processor architecture, availability of network mounted filesystems, etc.).”
- “Files saved on one node are available on all nodes.”
Content from Working with the scheduler
Last updated on 2025-01-14 | Edit this page
Estimated time: 80 minutes
Overview
Questions
- What is a scheduler and why are they used?
- How do I launch a program to run on any one node in the cluster?
- How do I capture the output of a program that is run on a node in the cluster?
Objectives
- Run a simple Hello World style program on the cluster.
- Submit a simple Hello World style script to the cluster.
- Use the batch system command line tools to monitor the execution of your job.
- Inspect the output and error files of your jobs.
Job Scheduler
An HPC system might have thousands of nodes and thousands of users. How do we decide who gets what and when? How do we ensure that a task is run with the resources it needs? This job is handled by a special piece of software called the scheduler. On an HPC system, the scheduler manages which jobs run where and when.
The following illustration compares these tasks of a job scheduler to a waiter in a restaurant. If you can relate to an instance where you had to wait for a while in a queue to get in to a popular restaurant, then you may now understand why sometimes your job do not start instantly as in your laptop.

The scheduler used in this lesson is Slurm. Although Slurm is not used everywhere, running jobs is quite similar regardless of what software is being used. The exact syntax might change, but the concepts remain the same.
Running a Batch Job
The most basic use of the scheduler is to run a command non-interactively. Any command (or series of commands) that you want to run on the cluster is called a job, and the process of using a scheduler to run the job is called batch job submission.
In this case, the job we want to run is just a shell script. Let’s
create a demo shell script to run as a test. The landing pad will have a
number of terminal-based text editors installed. Use whichever you
prefer. Unsure? nano is a pretty good, basic choice.
BASH
userid@ln03:~> nano example-job.sh
userid@ln03:~> chmod +x example-job.sh
userid@ln03:~> cat example-job.shOUTPUT
#!/bin/bash
echo -n "This script is running on "
hostnameOUTPUT
This script is running on ln03This job runs on the login node.
If you completed the previous challenge successfully, you probably
realise that there is a distinction between running the job through the
scheduler and just “running it”. To submit this job to the scheduler, we
use the sbatch command.
OUTPUT
sbatch: Warning: Your job has no time specification (--time=) and the default time is short. You can cancel your job with 'scancel <JOB_ID>' if you wish to resubmit.
sbatch: Warning: It appears your working directory may be on the home filesystem. It is /home2/home/ta133/ta133/userid. This is not available from the compute nodes - please check that this is what you intended. You can cancel your job with 'scancel <JOBID>' if you wish to resubmit.
Submitted batch job 286949Ah! What went wrong here? Slurm is telling us that the file system we
are currently on, /home, is not available on the compute
nodes and that we are getting the default, short runtime. We will deal
with the runtime later, but we need to move to a different file system
to submit the job and have it visible to the compute nodes. On ARCHER2,
this is the /work file system. The path is similar to home
but with /work at the start. Lets move there now, copy our
job script across and resubmit:
BASH
userid@ln03:~> cd /work/ta133/ta133/userid
userid@uan01:/work/ta158/ta158/userid> cp ~/example-job.sh .
userid@uan01:/work/ta158/ta158/userid> sbatch --partition=standard --qos=short example-job.shOUTPUT
Submitted batch job 36855That’s better! And that’s all we need to do to submit a job. Our work
is done — now the scheduler takes over and tries to run the job for us.
While the job is waiting to run, it goes into a list of jobs called the
queue. To check on our job’s status, we check the queue using
the command squeue -u userid.
OUTPUT
JOBID USER ACCOUNT NAME ST REASON START_TIME T...
36856 yourUsername yourAccount example-job.sh R None 2017-07-01T16:47:02 ...We can see all the details of our job, most importantly that it is in
the R or RUNNING state. Sometimes our jobs
might need to wait in a queue (PENDING) or have an error
(E).
The best way to check our job’s status is with squeue.
Of course, running squeue repeatedly to check on things can
be a little tiresome. To see a real-time view of our jobs, we can use
the watch command. watch reruns a given
command at 2-second intervals. This is too frequent, and will likely
upset your system administrator. You can change the interval to a more
reasonable value, for example 15 seconds, with the -n 15
parameter. Let’s try using it to monitor another job.
BASH
userid@uan01:/work/ta158/ta158/userid> sbatch --partition=standard --qos=short example-job.sh
userid@uan01:/work/ta158/ta158/userid> watch -n 15 squeue -u useridYou should see an auto-updating display of your job’s status. When it
finishes, it will disappear from the queue. Press Ctrl-c
when you want to stop the watch command.
Where’s the Output?
On the login node, this script printed output to the terminal — but
when we exit watch, there’s nothing. Where’d it go? HPC job
output is typically redirected to a file in the directory you launched
it from. Use ls to find and read the file.
Customising a Job
The job we just ran used some of the scheduler’s default options. In a real-world scenario, that’s probably not what we want. The default options represent a reasonable minimum. Chances are, we will need more cores, more memory, more time, among other special considerations. To get access to these resources we must customize our job script.
Comments in UNIX shell scripts (denoted by #) are
typically ignored, but there are exceptions. For instance the special
#! comment at the beginning of scripts specifies what
program should be used to run it (you’ll typically see
#!/bin/bash). Schedulers like Slurm also have a special
comment used to denote special scheduler-specific options. Though these
comments differ from scheduler to scheduler, Slurm’s special comment is
#SBATCH. Anything following the #SBATCH
comment is interpreted as an instruction to the scheduler.
Let’s illustrate this by example. By default, a job’s name is the
name of the script, but the --job-name option can be used
to change the name of a job. Add an option to the script:
OUTPUT
#!/bin/bash
#SBATCH --job-name new_name
echo -n "This script is running on "
hostname
echo "This script has finished successfully."Submit the job and monitor its status:
BASH
userid@uan01:/work/ta158/ta158/userid> sbatch --partition=standard --qos=short example-job.sh
userid@uan01:/work/ta158/ta158/userid> squeue -u useridOUTPUT
JOBID USER ACCOUNT NAME ST REASON START_TIME TIME TIME_LEFT NODES CPUS
38191 yourUsername yourAccount new_name PD Priority N/A 0:00 1:00:00 1 1Fantastic, we’ve successfully changed the name of our job!
Resource Requests
But what about more important changes, such as the number of cores and memory for our jobs? One thing that is absolutely critical when working on an HPC system is specifying the resources required to run a job. This allows the scheduler to find the right time and place to schedule our job. If you do not specify requirements (such as the amount of time you need), you will likely be stuck with your site’s default resources, which is probably not what you want.
The following are several key resource requests:
-
--nodes=<nodes>- Number of nodes to use -
--ntasks-per-node=<tasks-per-node>- Number of parallel processes per node -
--cpus-per-task=<cpus-per-task>- Number of cores to assign to each parallel process -
--time=<days-hours:minutes:seconds>- Maximum real-world time (walltime) your job will be allowed to run. The<days>part can be omitted.
Note that just requesting these resources does not make your job run faster, nor does it necessarily mean that you will consume all of these resources. It only means that these are made available to you. Your job may end up using less memory, or less time, or fewer tasks or nodes, than you have requested, and it will still run.
It’s best if your requests accurately reflect your job’s requirements. We’ll talk more about how to make sure that you’re using resources effectively in a later episode of this lesson.
Command line options or job script options?
All of the options we specify can be supplied on the command line (as
we do here for --partition=standard) or in the job script
(as we have done for the job name above). These are interchangeable. It
is often more convenient to put the options in the job script as it
avoids lots of typing at the command line.
Submitting Resource Requests
Modify our hostname script so that it runs for a minute,
then submit a job for it on the cluster. You should also move all the
options we have been specifying on the command line
(e.g. --partition) into the script at this point.
OUTPUT
#!/bin/bash
#SBATCH --time 00:01:15
#SBATCH --partition=standard
#SBATCH --qos=short
echo -n "This script is running on "
sleep 60 # time in seconds
hostname
echo "This script has finished successfully."Why are the Slurm runtime and sleep time not
identical?
Job environment variables
When Slurm runs a job, it sets a number of environment variables for
the job. One of these will let us check our work from the last problem.
The SLURM_CPUS_PER_TASK variable is set to the number of
CPUs we requested with -c. Using the
SLURM_CPUS_PER_TASK variable, modify your job so that it
prints how many CPUs have been allocated.
Resource requests are typically binding. If you exceed them, your job will be killed. Let’s use walltime as an example. We will request 30 seconds of walltime, and attempt to run a job for two minutes.
OUTPUT
#!/bin/bash
#SBATCH --job-name long_job
#SBATCH --time 00:00:30
#SBATCH --partition=standard
#SBATCH --qos=short
echo "This script is running on ... "
sleep 120 # time in seconds
hostname
echo "This script has finished successfully."Submit the job and wait for it to finish. Once it is has finished, check the log file.
BASH
userid@uan01:/work/ta158/ta158/userid> sbatch example-job.sh
userid@uan01:/work/ta158/ta158/userid> watch -n 15 squeue -u useridOUTPUT
This job is running on:
nid001147
slurmstepd: error: *** JOB 38193 ON cn01 CANCELLED AT 2017-07-02T16:35:48 DUE TO TIME LIMIT ***Our job was killed for exceeding the amount of resources it requested. Although this appears harsh, this is actually a feature. Strict adherence to resource requests allows the scheduler to find the best possible place for your jobs. Even more importantly, it ensures that another user cannot use more resources than they’ve been given. If another user messes up and accidentally attempts to use all of the cores or memory on a node, Slurm will either restrain their job to the requested resources or kill the job outright. Other jobs on the node will be unaffected. This means that one user cannot mess up the experience of others, the only jobs affected by a mistake in scheduling will be their own.
But how much does it cost?
Although your job will be killed if it exceeds the selected runtime, a job that completes within the time limit is only charged for the time it actually used. However, you should always try and specify a wallclock limit that is close to (but greater than!) the expected runtime as this will enable your job to be scheduled more quickly. If you say your job will run for an hour, the scheduler has to wait until a full hour becomes free on the machine. If it only ever runs for 5 minutes, you could have set a limit of 10 minutes and it might have been run earlier in the gaps between other users’ jobs.
Cancelling a Job
Sometimes we’ll make a mistake and need to cancel a job. This can be
done with the scancel command. Let’s submit a job and then
cancel it using its job number (remember to change the walltime so that
it runs long enough for you to cancel it before it is killed!).
BASH
userid@uan01:/work/ta158/ta158/userid> sbatch example-job.sh
userid@uan01:/work/ta158/ta158/userid> squeue -u useridOUTPUT
Submitted batch job 38759
JOBID USER ACCOUNT NAME ST REASON START_TIME TIME TIME_LEFT NODES CPUS
38759 yourUsername yourAccount example-job.sh PD Priority N/A 0:00 1:00 1 1Now cancel the job with its job number (printed in your terminal). Absence of any job info indicates that the job has been successfully cancelled.
BASH
userid@uan01:/work/ta158/ta158/userid> scancel 38759
# It might take a minute for the job to disappear from the queue...
userid@uan01:/work/ta158/ta158/userid> squeue -u useridOUTPUT
JOBID USER ACCOUNT NAME ST REASON START_TIME TIME TIME_LEFT NODES CPUSCancelling multiple jobs
We can also cancel all of our jobs at once using the -u
option. This will delete all jobs for a specific user (in this case us).
Note that you can only delete your own jobs. Try submitting multiple
jobs and then cancelling them all with
scancel -u yourUsername.
Other Types of Jobs
Up to this point, we’ve focused on running jobs in batch mode. Slurm also provides the ability to start an interactive session.
There are very frequently tasks that need to be done interactively.
Creating an entire job script might be overkill, but the amount of
resources required is too much for a login node to handle. A good
example of this might be building a genome index for alignment with a
tool like HISAT2.
Fortunately, we can run these types of tasks as a one-off with
srun.
srun runs a single command in the queue system and then
exits. Let’s demonstrate this by running the hostname
command with srun. (We can cancel an srun job
with Ctrl-c.)
OUTPUT
nid001976srun accepts all of the same options as
sbatch. However, instead of specifying these in a script,
these options are specified on the command-line when starting a job.
Typically, the resulting shell environment will be the same as that
for sbatch.
Interactive jobs
Sometimes, you will need a lot of resource for interactive use.
Perhaps it’s our first time running an analysis or we are attempting to
debug something that went wrong with a previous job. Fortunately, SLURM
makes it easy to start an interactive job with srun:
You should be presented with a bash prompt. Note that the prompt may
change to reflect your new location, in this case the compute node we
are logged on. You can also verify this with hostname.
When you are done with the interactive job, type exit to
quit your session.
Running parallel jobs using MPI
As we have already seen, the power of HPC systems comes from parallelism, i.e. having lots of processors/disks etc. connected together rather than having more powerful components than your laptop or workstation. Often, when running research programs on HPC you will need to run a program that has been built to use the MPI (Message Passing Interface) parallel library. The MPI library allows programs to exploit multiple processing cores in parallel to allow researchers to model or simulate faster on larger problem sizes. The details of how MPI work are not important for this course or even to use programs that have been built using MPI; however, MPI programs typically have to be launched in job submission scripts in a different way to serial programs and users of parallel programs on HPC systems need to know how to do this. Specifically, launching parallel MPI programs typically requires four things:
- A special parallel launch program such as
mpirun,mpiexec,srunoraprun. - A specification of how many processes to use in parallel. For example, our parallel program may use 256 processes in parallel.
- A specification of how many parallel processes to use per compute node. For example, if our compute nodes each have 32 cores we often want to specify 32 parallel processes per node.
- The command and arguments for our parallel program.
Required Files
The program used in this example can be retrieved using wget or a browser and copied to the remote.
Using wget:
Using a web browser:
https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/pi-mpi.py
To illustrate this process, we will use a simple MPI parallel program
that estimates the value of Pi. (We will meet this example program in
more detail in a later episode.) Here is a job submission script that
runs the program across two compute nodes on the cluster. Create a file
(e.g. called: run-pi-mpi.slurm) with the contents of this
script in it.
BASH
#!/bin/bash
#SBATCH --partition=standard
#SBATCH --qos=short
#SBATCH --time=00:05:00
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=16
module load cray-python
srun python pi-mpi.py 10000000The parallel launch line for our program can be seen towards the bottom of the script:
And this corresponds to the four required items we described above:
- Parallel launch program: in this case the parallel launch program is
called
srun; the additional argument controls which cores are used. - Number of parallel processes per node: in this case this is 16, and
is specified by the option
--ntasks-per-node=16option. - Total number of parallel processes: in this case this is also 16, because we specified 1 node and 16 parallel processes per node.
- Our program and arguments: in this case this is
python pi-mpi.py 10000000.
As for our other jobs, we launch using the sbatch
command.
The program generates no output with all details printed to the job log.
Running parallel jobs
Modify the pi-mpi-run script that you used above to use all 128 cores on one node. Check the output to confirm that it used the correct number of cores in parallel for the calculation.
Configuring parallel jobs
You will see in the job output that information is displayed about where each MPI process is running, in particular which node it is on.
Modify the pi-mpi-run script that you run a total of 2 nodes and 16 processes; but to use only 8 tasks on each of two nodes. Check the output file to ensure that you understand the job distribution.
Key Points
- “The scheduler handles how compute resources are shared between users.”
- “Everything you do should be run through the scheduler.”
- “A job is just a shell script.”
- “If in doubt, request more resources than you will need.”
Content from Accessing software via Modules
Last updated on 2025-01-14 | Edit this page
Estimated time: 45 minutes
Overview
Questions
- How do we load and unload software packages?
Objectives
- Understand how to load and use a software package.
On a high-performance computing system, it is seldom the case that the software we want to use is available when we log in. It is installed, but we will need to “load” it before it can run.
Before we start using individual software packages, however, we should understand the reasoning behind this approach. The three biggest factors are:
- software incompatibilities
- versioning
- dependencies
Software incompatibility is a major headache for programmers.
Sometimes the presence (or absence) of a software package will break
others that depend on it. Two of the most famous examples are Python 2
and 3 and C compiler versions. Python 3 famously provides a
python command that conflicts with that provided by Python
2. Software compiled against a newer version of the C libraries and then
used when they are not present will result in a nasty
'GLIBCXX_3.4.20' not found error, for instance.
Software versioning is another common issue. A team might depend on a certain package version for their research project - if the software version was to change (for instance, if a package was updated), it might affect their results. Having access to multiple software versions allow a set of researchers to prevent software versioning issues from affecting their results.
Dependencies are where a particular software package (or even a particular version) depends on having access to another software package (or even a particular version of another software package). For example, the VASP materials science software may depend on having a particular version of the FFTW (Fastest Fourier Transform in the West) software library available for it to work.
Environment Modules
Environment modules are the solution to these problems. A module is a self-contained description of a software package — it contains the settings required to run a software package and, usually, encodes required dependencies on other software packages.
There are a number of different environment module implementations
commonly used on HPC systems: the two most common are TCL
modules and Lmod. Both of these use similar syntax and the
concepts are the same so learning to use one will allow you to use
whichever is installed on the system you are using. In both
implementations the module command is used to interact with
environment modules. An additional subcommand is usually added to the
command to specify what you want to do. For a list of subcommands you
can use module -h or module help. As for all
commands, you can access the full help on the man pages with
man module.
On login you may start out with a default set of modules loaded or you may start out with an empty environment; this depends on the setup of the system you are using.
Listing Available Modules
To see available software modules, use module avail:
OUTPUT
----------- /work/y07/shared/archer2-modules/modulefiles-cse-pyvenvs -----------
tensorflow/2.3.1-py38 torch/1.6.0-py38
----------- /work/y07/shared/archer2-modules/modulefiles-cse-pymods ------------
python-netCDF4/1.5.5.1
------------ /work/y07/shared/archer2-modules/modulefiles-cse-utils ------------
bolt/0.7 ncview/ncview-2.1.7-gcc-10.1.0 vmd/1.9.3-mpi-gcc10
cmake/3.18.4 reframe/3.2 xios/2.5-gcc10
ed/1.16-gcc10 tcl/8.4.20-gcc10 xthi/1.0
epcc-job-env tcl/8.5.0-gcc10 xthi/1.0-gcc10
epcc-reframe/0.1 tcl/8.6.0-gcc10
genmaskcpu/1.0 tcl/8.6.10-gcc10(default)
gnuplot/5.4.1-gcc-10.1.0 tk/8.5.6-gcc10
lzip/1.20-gcc10 tk/8.6.10-gcc10(default)
nco/4.9.6 visidata/2.1
nco/4.9.6-gcc-10.1.0 vmd/1.9.3-gcc10(default)
------------ /work/y07/shared/archer2-modules/modulefiles-cse-libs -------------
adios/1.13.1 hypre/2.18.0 mumps/5.2.1 superlu-dist/6.1.1
boost/1.72.0 libxml2/2.9.7-gcc-9.3.0 parmetis/4.0.3 superlu/5.2.1
glm/0.9.9.6 matio/1.5.18 petsc/3.13.3 trilinos/12.18.1
gmp/6.1.2-gcc10 metis/5.1.0 scotch/6.0.10
...Listing Currently Loaded Modules
You can use the module list command to see which modules
you currently have loaded in your environment. If you have no modules
loaded, you will see a message telling you so
OUTPUT
Currently Loaded Modulefiles:
1) cpe-cray 8) perftools-base/20.10.0(default)
2) cce/10.0.4(default) 9) xpmem/2.2.35-7.0.1.0_1.9__gd50fabf.shasta(default)
3) craype/2.7.2(default) 10) cray-mpich/8.0.16(default)
4) craype-x86-rome 11) cray-libsci/20.10.1.2(default)
5) libfabric/1.11.0.0.233(default) 12) bolt/0.7
6) craype-network-ofi 13) /work/y07/shared/archer2-modules/modulefiles-cse/epcc-setup-env
7) cray-dsmml/0.1.2(default) 14) /usr/local/share/epcc-module/epcc-module-loader Loading and Unloading Software
To load a software module, use module load. Let’s say we
would like to use the NetCDF utility ncdump.
On login, ncdump is not available. We can test this by
using the which command. which looks for
programs the same way that Bash does, so we can use it to tell us where
a particular piece of software is stored.
OUTPUT
which: no ncdump in (/usr/local/maven/bin:/lus/cls01095/work/y07/shared/bolt/0.7/bin:/work/y07/shared/utils/bin:/opt/cray/pe/perftools/20.10.0/bin:/opt/cray/pe/papi/6.0.0.4/bin:/opt/cray/libfabric/1.11.0.0.233/bin:/opt/cray/pe/craype/2.7.2/bin:/opt/cray/pe/cce/10.0.4/cce-clang/x86_64/bin:/opt/cray/pe/cce/10.0.4/binutils/x86_64/x86_64-pc-linux-gnu/bin:/opt/cray/pe/cce/10.0.4/binutils/cross/x86_64-aarch64/aarch64-linux-gnu/../bin:/opt/cray/pe/cce/10.0.4/utils/x86_64/bin:/usr/local/Modules/bin:/usr/local/bin:/usr/bin:/bin:/opt/cray/pe/bin:/usr/lib/mit/bin)We can find the ncdump command by using
module load:
OUTPUT
/opt/cray/pe/netcdf/4.7.4.2/bin/ncdumpSo, what just happened?
To understand the output, first we need to understand the nature of
the $PATH environment variable. $PATH is a
special environment variable that controls where a UNIX system looks for
software. Specifically, $PATH is a list of directories
(separated by :) that the OS searches through for a command
before giving up and telling us it can’t find it. As with all
environment variables we can print it out using echo.
OUTPUT
/opt/cray/pe/netcdf/4.7.4.2/bin:/opt/cray/pe/python/3.8.5.0/bin:/lus/cls01095/work/z19/z19/aturner/.local/bin:/lus/cls01095/work/y07/shared/bolt/0.7/bin:/work/y07/shared/utils/bin:/usr/local/maven/bin:/opt/cray/pe/perftools/20.10.0/bin:/opt/cray/pe/papi/6.0.0.4/bin:/opt/cray/libfabric/1.11.0.0.233/bin:/opt/cray/pe/craype/2.7.2/bin:/opt/cray/pe/cce/10.0.4/cce-clang/x86_64/bin:/opt/cray/pe/cce/10.0.4/binutils/x86_64/x86_64-pc-linux-gnu/bin:/opt/cray/pe/cce/10.0.4/binutils/cross/x86_64-aarch64/aarch64-linux-gnu/../bin:/opt/cray/pe/cce/10.0.4/utils/x86_64/bin:/usr/local/Modules/bin:/home/z19/z19/aturner/bin:/usr/local/bin:/usr/bin:/bin:/opt/cray/pe/bin:/usr/lib/mit/binYou’ll notice a similarity to the output of the which
command. In this case, there’s only one difference: the different
directory at the beginning. When we ran the module load
command, it added a directory to the beginning of our
$PATH. Let’s examine what’s there:
OUTPUT
nc-config nccopy ncdump ncgen ncgen3 ncxx4-config nf-configIn summary, module load will add software to your
$PATH. module load may also load additional
modules with software dependencies.
To unload a module, use module unload with the relevant
module name.
Unload!
Confirm you can unload the cray-netcdf module and check
what happens to the PATH environment variable.
Software versioning
So far, we’ve learned how to load and unload software packages. This is very useful. However, we have not yet addressed the issue of software versioning. At some point or other, you will run into issues where only one particular version of some software will be suitable. Perhaps a key bugfix only happened in a certain version, or version X broke compatibility with a file format you use. In either of these example cases, it helps to be very specific about what software is loaded.
Let’s examine the output of module avail more
closely.
OUTPUT
--------------------------- /opt/cray/pe/modulefiles ---------------------------
cray-netcdf-hdf5parallel/4.7.4.0 cray-netcdf/4.7.4.0
cray-netcdf-hdf5parallel/4.7.4.2(default) cray-netcdf/4.7.4.2(default) Note that we have two different versions of cray-netcdf
(and also two versions of something else
cray-netcdf-hdf5parallel which match our search).
Using module swap
Load module cray-netcdf as before. Note that if we do
not specifify a particular version, we load a default version. If we
wish to change versions, we can use
module swap <old-module> <new-module>. Try this
to obtain cray-netcdf/4.7.4.0. Check what has happened to
the location of the ncdump utility.
Using Software Modules in Scripts
Create a job that is able to run ncdump --version.
Running a job is just like logging on to the system (you should not
assume a module loaded on the login node is loaded on a compute
node).
Key Points
- “Load software with
module load softwareName.” - “Unload software with
module purge” - “The module system handles software versioning and package conflicts for you automatically.”
Content from Transferring files with remote computers
Last updated on 2025-01-14 | Edit this page
Estimated time: 30 minutes
Overview
Questions
- How do I transfer files to (and from) the cluster?
Objectives
-
wgetandcurl -Odownload a file from the internet. -
scptransfers files to and from your computer.
Required Files
The program used in this example can be retrieved using wget or a browser and copied to the remote.
Using wget:
BASH
userid@ln03:~> wget https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gzUsing a web browser:
https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gz
Computing with a remote computer offers very limited use if we cannot get files to or from the cluster. There are several options for transferring data between computing resources, from command line options to GUI programs, which we will cover here.
Download Files From the Internet
One of the most straightforward ways to download files is to use
either curl or wget, one of these is usually
installed in most Linux shells, on Mac OS terminal and in GitBash. Any
file that can be downloaded in your web browser through a direct link
can be downloaded using curl -O or wget. This
is a quick way to download datasets or source code.
The syntax for these commands is:
curl -O https://some/link/to/a/file and
wget https://some/link/to/a/file. Try it out by downloading
some material we’ll use later on, from a terminal on your local
machine.
BASH
[user@laptop ~]$ curl -O https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gzor
BASH
[user@laptop ~]$ wget https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gz
tar.gz?
This is an archive file format, just like .zip, commonly
used and supported by default on Linux, which is the operating system
the majority of HPC cluster machines run. You may also see the extension
.tgz, which is exactly the same. We’ll talk more about
“tarballs,” since “tar-dot-g-z” is a mouthful, later on.
Transferring Single Files and Folders With scp
To copy a single file to or from the cluster, we can use
scp (“secure copy”). The syntax can be a little complex for
new users, but we’ll break it down.
To upload to another computer:
To download from another computer:
Note that everything after the : is relative to our home
directory on the remote computer. We can leave it at that if we don’t
care where the file goes.
Upload a File
Copy the file you just downloaded from the Internet to your home directory on ARCHER2.
Why Not Download on ARCHER2 Directly?
Some computer clusters are behind firewalls set to only allow
transfers initiated from the outside. This means that the
curl command will fail, as an address outside the firewall
is unreachable from the inside. To get around this, run the
curl or wget command from your local machine
to download the file, then use the scp command (just below
here) to upload it to the cluster.
Can you download from the server directly?
Try downloading the file directly using curl or
wget. Do the commands understand file locations on your
local machine over SSH? Note that it may well fail, and that’s OK!
Using curl or wget commands like the
following:
BASH
[user@laptop ~]$ ssh userid@login.archer2.ac.uk
userid@ln03:~> curl -O https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gz
or
userid@ln03:~> wget https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/hpc-intro-data.tar.gzDid it work? If not, what does the terminal output tell you about what happened?
To copy a whole directory, we add the -r flag, for
“recursive”: copy the item specified, and every item
below it, and every item below those… until it reaches the bottom of the
directory tree rooted at the folder name you provided.
Caution
For a large directory — either in size or number of files — copying
with -r can take a long time to complete.
What’s in a /?
When using scp, you may have noticed that a
: always follows the remote computer name;
sometimes a / follows that, and sometimes not, and
sometimes there’s a final /. On Linux computers,
/ is the root directory, the
location where the entire filesystem (and others attached to it) is
anchored. A path starting with a / is called
absolute, since there can be nothing above the root
/. A path that does not start with / is called
relative, since it is not anchored to the root.
If you want to upload a file to a location inside your home directory
— which is often the case — then you don’t need a leading
/. After the :, start writing the sequence of
folders that lead to the final storage location for the file or, as
mentioned above, provide nothing if your home directory is the
destination.
A trailing slash on the target directory is optional, and has no
effect for scp -r, but is important in other commands, like
rsync.
A Note on rsync
As you gain experience with transferring files, you may find the
scp command limiting. The rsync utility provides advanced
features for file transfer and is typically faster compared to both
scp and sftp (see below). It is especially
useful for transferring large and/or many files and creating synced
backup folders. The syntax is similar to scp. To transfer
to another computer with commonly used options:
BASH
[user@laptop ~]$ rsync -avzP path/to/local/file.txt userid@login.archer2.ac.uk:directory/path/on/ARCHER2/The a (archive) option preserves file timestamps and
permissions among other things; the v (verbose) option
gives verbose output to help monitor the transfer; the z
(compression) option compresses the file during transit to reduce size
and transfer time; and the P (partial/progress) option
preserves partially transferred files in case of an interruption and
also displays the progress of the transfer.
To recursively copy a directory, we can use the same options:
BASH
[user@laptop ~]$ rsync -avzP path/to/local/dir userid@login.archer2.ac.uk:directory/path/on/ARCHER2/As written, this will place the local directory and its contents under the specified directory on the remote system. If the trailing slash is omitted on the destination, a new directory corresponding to the transferred directory (‘dir’ in the example) will not be created, and the contents of the source directory will be copied directly into the destination directory.
The a (archive) option implies recursion.
To download a file, we simply change the source and destination:
A Note on Ports
All file transfers using the above methods use SSH to encrypt data
sent through the network. So, if you can connect via SSH, you will be
able to transfer files. By default, SSH uses network port 22. If a
custom SSH port is in use, you will have to specify it using the
appropriate flag, often -p, -P, or
--port. Check --help or the man
page if you’re unsure.
Archiving Files
One of the biggest challenges we often face when transferring data between remote HPC systems is that of large numbers of files. There is an overhead to transferring each individual file and when we are transferring large numbers of files these overheads combine to slow down our transfers to a large degree.
The solution to this problem is to archive multiple files into smaller numbers of larger files before we transfer the data to improve our transfer efficiency. Sometimes we will combine archiving with compression to reduce the amount of data we have to transfer and so speed up the transfer.
The most common archiving command you will use on a (Linux) HPC
cluster is tar. tar can be used to combine
files into a single archive file and, optionally, compress it.
Let’s start with the file we downloaded from the lesson site,
hpc-lesson-data.tar.gz. The “gz” part stands for
gzip, which is a compression library. Reading this file name,
it appears somebody took a folder named “hpc-lesson-data,” wrapped up
all its contents in a single file with tar, then compressed
that archive with gzip to save space. Let’s check using
tar with the -t flag, which prints the
“table of contents” without unpacking the file,
specified by -f <filename>, on the remote computer.
Note that you can concatenate the two flags, instead of writing
-t -f separately.
BASH
[user@laptop ~]$ ssh userid@login.archer2.ac.uk
userid@ln03:~> tar -tf hpc-lesson-data.tar.gz
hpc-intro-data/
hpc-intro-data/north-pacific-gyre/
hpc-intro-data/north-pacific-gyre/NENE01971Z.txt
hpc-intro-data/north-pacific-gyre/goostats
hpc-intro-data/north-pacific-gyre/goodiff
hpc-intro-data/north-pacific-gyre/NENE02040B.txt
hpc-intro-data/north-pacific-gyre/NENE01978B.txt
hpc-intro-data/north-pacific-gyre/NENE02043B.txt
hpc-intro-data/north-pacific-gyre/NENE02018B.txt
hpc-intro-data/north-pacific-gyre/NENE01843A.txt
hpc-intro-data/north-pacific-gyre/NENE01978A.txt
hpc-intro-data/north-pacific-gyre/NENE01751B.txt
hpc-intro-data/north-pacific-gyre/NENE01736A.txt
hpc-intro-data/north-pacific-gyre/NENE01812A.txt
hpc-intro-data/north-pacific-gyre/NENE02043A.txt
hpc-intro-data/north-pacific-gyre/NENE01729B.txt
hpc-intro-data/north-pacific-gyre/NENE02040A.txt
hpc-intro-data/north-pacific-gyre/NENE01843B.txt
hpc-intro-data/north-pacific-gyre/NENE01751A.txt
hpc-intro-data/north-pacific-gyre/NENE01729A.txt
hpc-intro-data/north-pacific-gyre/NENE02040Z.txtThis shows a folder containing another folder, which contains a bunch
of files. If you’ve taken The Carpentries’ Shell lesson recently, these
might look familiar. Let’s see about that compression, using
du for “disk usage”.
Files Occupy at Least One “Block”
If the filesystem block size is larger than 36 KB, you’ll see a larger number: files cannot be smaller than one block.
Now let’s unpack the archive. We’ll run tar with a few
common flags:
-
-xto extract the archive -
-vfor verbose output -
-zfor gzip compression -
-ffor the file to be unpacked
When it’s done, check the directory size with du and
compare.
Extract the Archive
Using the four flags above, unpack the lesson data using
tar. Then, check the size of the whole unpacked directory
using du. Hint: tar lets you concatenate
flags.
OUTPUT
hpc-intro-data/
hpc-intro-data/north-pacific-gyre/
hpc-intro-data/north-pacific-gyre/NENE01971Z.txt
hpc-intro-data/north-pacific-gyre/goostats
hpc-intro-data/north-pacific-gyre/goodiff
hpc-intro-data/north-pacific-gyre/NENE02040B.txt
hpc-intro-data/north-pacific-gyre/NENE01978B.txt
hpc-intro-data/north-pacific-gyre/NENE02043B.txt
hpc-intro-data/north-pacific-gyre/NENE02018B.txt
hpc-intro-data/north-pacific-gyre/NENE01843A.txt
hpc-intro-data/north-pacific-gyre/NENE01978A.txt
hpc-intro-data/north-pacific-gyre/NENE01751B.txt
hpc-intro-data/north-pacific-gyre/NENE01736A.txt
hpc-intro-data/north-pacific-gyre/NENE01812A.txt
hpc-intro-data/north-pacific-gyre/NENE02043A.txt
hpc-intro-data/north-pacific-gyre/NENE01729B.txt
hpc-intro-data/north-pacific-gyre/NENE02040A.txt
hpc-intro-data/north-pacific-gyre/NENE01843B.txt
hpc-intro-data/north-pacific-gyre/NENE01751A.txt
hpc-intro-data/north-pacific-gyre/NENE01729A.txt
hpc-intro-data/north-pacific-gyre/NENE02040Z.txtNote that we did not type out -x -v -z -f, thanks to the
flag concatenation, though the command works identically either way.
Was the Data Compressed?
Text files compress nicely: the “tarball” is one-quarter the total size of the raw data!
If you want to reverse the process — compressing raw data instead of
extracting it — set a c flag instead of x, set
the archive filename, then provide a directory to compress:
Working with Windows
When you transfer text files to from a Windows system to a Unix
system (Mac, Linux, BSD, Solaris, etc.) this can cause problems. Windows
encodes its files slightly different than Unix, and adds an extra
character to every line. On a Unix system, every line in a file ends
with a \n (newline). On Windows, every line in a file ends
with a \r\n (carriage return + newline). This causes
problems sometimes. Though most modern programming languages and
software handles this correctly, in some rare instances, you may run
into an issue. The solution is to convert a file from Windows to Unix
encoding with the dos2unix command. You can identify if a
file has Windows line endings with cat -A filename. A file
with Windows line endings will have ^M$ at the end of every
line. A file with Unix line endings will have $ at the end
of a line. To convert the file, just run dos2unix filename.
(Conversely, to convert back to Windows format, you can run
unix2dos filename.)
Key Points
- “
wgetandcurl -Odownload a file from the internet.” - “
scptransfers files to and from your computer.”
Content from Using resources effectively
Last updated on 2025-01-14 | Edit this page
Estimated time: 40 minutes
Overview
Questions
- How do we monitor our jobs?
- How can I get my jobs scheduled more easily?
Objectives
- Understand how to look up job statistics and profile code.
- Understand job size implications.
We’ve touched on all the skills you need to interact with an HPC cluster: logging in over SSH, loading software modules, submitting parallel jobs, and finding the output. Let’s learn about estimating resource usage and why it might matter. To do this we need to understand the basics of benchmarking. Benchmarking is essentially performing simple experiments to help understand how the performance of our work varies as we change the properties of the jobs on the cluster - including input parameters, job options and resources used.
Our example
In the rest of this episode, we will use an example parallel application that calculates an estimate of the value of Pi. Although this is a toy problem, it exhibits all the properties of a full parallel application that we are interested in for this course.
The main resource we will consider here is the use of compute core time as this is the resource you are usually charged for on HPC resources. However, other resources - such as memory use - may also have a bearing on how you choose resources and constrain your choice.
For those that have come across HPC benchmarking before, you may be aware that people often make a distinction between strong scaling and weak scaling:
- Strong scaling is where the problem size (i.e. the application) stays the same size and we try to use more cores to solve the problem faster.
- Weak scaling is where the problem size increases at the same rate as we increase the core count so we are using more cores to solve a larger problem.
Both of these approaches are equally valid uses of HPC. This example looks at strong scaling.
Before we work on benchmarking, it is useful to define some terms for the example we will be using
-
Program The computer program we are executing
(
pi-mpi.pyin the examples below) - Application The combination of computer program with particular input parameters
Accessing the software and input
Required Files
The program used in this example can be retrieved using wget or a browser and copied to the remote.
Using wget:
Using a web browser:
https://epcced.github.io/2024-06-20-hpc-intro-shampton/files/pi-mpi.py
Baseline: running in serial
Before starting to benchmark an application to understand what resources are best to use, you need a baseline performance result. In more formal benchmarking, your baseline is usually the minimum number of cores or nodes you can run on. However, for understanding how best to use resources, as we are doing here, your baseline could be the performance on any number of cores or nodes that you can measure the change in performance from.
Our pi-mpi.py application is small enough that we can
run a serial (i.e. using a single core) job for our baseline performance
so that is where we will start
Run a single core job
Write a job submission script that runs the pi-mpi.py
application on a single core. You will need to take an initial guess as
to the walltime to request to give the job time to complete. Submit the
job and check the contents of the STDOUT file to see if the application
worked or not.
Creating a file called submit-pi-mpi.slurm:
BASH
#!/bin/bash
#SBATCH --partition=standard
#SBATCH --qos=short
#SBATCH --job-name=pi-mpi
#SBATCH --nodes=1
#SBATCH --tasks-per-node=1
#SBATCH --time=00:15:00
srun python pi-mpi.py 10000000Run application using a single process (i.e. in serial) with a
blocking srun command:
Submit with to the batch queue with:
Output in the job log should look something like:
OUTPUT
Generating 10000000 samples.
Rank 0 generating 10000000 samples on host nid001246.
Numpy Pi: 3.141592653589793
My Estimate of Pi: 3.1416708
1 core(s), 10000000 samples, 228.881836 MiB memory, 0.423903 seconds, -0.002487% errorOnce your job has run, you should look in the output to identify the
performance. Most HPC programs should print out timing or performance
information (usually somewhere near the bottom of the summary output)
and pi-mpi.py is no exception. You should see two lines in
the output that look something like:
BASH
256 core(s), 100000000 samples, 2288.818359 MiB memory, 0.135041 seconds, -0.004774% error
Total run time=0.18654435999997077sYou can also get an estimate of the overall run time from the final
job statistics. If we look at how long the finished job ran for, this
will provide a quick way to see roughly what the runtime was. This can
be useful if you want to know quickly if a job was faster or not than a
previous job (as you do not have to find the output file to look up the
performance) but the number is not as accurate as the performance
recorded by the application itself and also includes static overheads
from running the job (such as loading modules and startup time) that can
skew the timings. To do this on use sacct -l -j with the
job ID, e.g.:
OUTPUT
JOBID USER ACCOUNT NAME ST REASON START_TIME T...
36856 yourUsername yourAccount example-job.sh R None 2017-07-01T16:47:02 ...Running in parallel and benchmarking performance
We have now managed to run the pi-mpi.py application
using a single core and have a baseline performance we can use to judge
how well we are using resources on the system.
Note that we also now have a good estimate of how long the application takes to run so we can provide a better setting for the walltime for future jobs we submit. Lets now look at how the runtime varies with core count.
Benchmarking the parallel performance
Modify your job script to run on multiple cores and evaluate the
performance of pi-mpi.py on a variety of different core
counts and use multiple runs to complete a table like the one below. If
you examine the log file you will see that it contains two timings: the
total time taken by the entire program and the time taken solely by the
calculation. The calculation of Pi from the Monte-Carlo counts is not
parallelised so this is a serial overhead, performed by a single
processor. The calculation part is, in theory, perfectly parallel (each
processor operates on independent sets of unique random numbers ) so
this should get faster on more cores. The Calculation core seconds is
the calculation time multiplied by the number of cores.
| Cores | Overall run time (s) | Calculation time (s) | Calculation core seconds |
|---|---|---|---|
| 1 (serial) | |||
| 2 | |||
| 4 | |||
| 8 | |||
| 16 | |||
| 32 | |||
| 64 | |||
| 128 | |||
| 256 |
Look at your results – do they make sense? Given the structure of the code, you would expect the performance of the calculation to increase linearly with the number of cores: this would give a roughly constant figure for the Calculation core seconds. Is this what you observe?
The table below shows example timings for runs on ARCHER2
| Cores | Overall run time (s) | Calculation time (s) | Calculation core seconds |
|---|---|---|---|
| 1 | 3.931 | 3.854 | 3.854 |
| 2 | 2.002 | 1.930 | 3.859 |
| 4 | 1.048 | 0.972 | 3.888 |
| 8 | 0.572 | 0.495 | 3.958 |
| 16 | 0.613 | 0.536 | 8.574 |
| 32 | 0.360 | 0.278 | 8.880 |
| 64 | 0.249 | 0.163 | 10.400 |
| 128 | 0.170 | 0.083 | 10.624 |
| 256 | 0.187 | 0.135 | 34.560 |
Understanding the performance
Now we have some data showing the performance of our application we need to try and draw some useful conclusions as to what the most efficient set of resources are to use for our jobs. To do this we introduce two metrics:
- Actual speedup The ratio of the baseline runtime (or runtime on the lowest core count) to the runtime at the specified core count. i.e. baseline runtime divided by runtime at the specified core count.
- Ideal speedup The expected speedup if the application showed perfect scaling. i.e. if you double the number of cores, the application should run twice as fast.
- Parallel efficiency The fraction of ideal speedup actually obtained for a given core count. This gives an indication of how well you are exploiting the additional resources you are using.
We will now use our performance results to compute these two metrics for the sharpen application and use the metrics to evaluate the performance and make some decisions about the most effective use of resources.
Computing the speedup and parallel efficiency
Use your Overall run times from above to fill in a table like the one below.
| Cores | Overall run time (s) | Actual speedup | Ideal speedup | Parallel efficiency |
|---|---|---|---|---|
| 1 (serial) | \(t_{c1}\) | - | 1 | 1 |
| 2 | \(t_{c2}\) | \(s_2 = t_{c1}/t_{c2}\) | \(i_2 = 2\) | \(s_2 / i_2\) |
| 4 | \(t_{c4}\) | \(s_4 = t_{c1}/t_{c4}\) | \(i_4 = 4\) | \(s_4 / i_4\) |
| 8 | ||||
| 16 | ||||
| 32 | ||||
| 64 | ||||
| 128 | ||||
| 256 |
Given your results, try to answer the following questions:
- What is the core count where you get the most efficient use of resources, irrespective of run time?
- What is the core count where you get the fastest solution, irrespective of efficiency?
- What do you think a good core count choice would be for this application that balances time to solution and efficiency? Why did you choose this option?
The table below gives example results for ARCHER2 based on the example runtimes given in the solution above.
| Cores | Overall run time (s) | Actual speedup | Ideal speedup | Parallel efficiency |
|---|---|---|---|---|
| 1 | 3.931 | 1.000 | 1.000 | 1.000 |
| 2 | 2.002 | 1.963 | 2.000 | 0.982 |
| 4 | 1.048 | 3.751 | 4.000 | 0.938 |
| 8 | 0.572 | 6.872 | 8.000 | 0.859 |
| 16 | 0.613 | 6.408 | 16.000 | 0.401 |
| 32 | 0.360 | 10.928 | 32.000 | 0.342 |
| 64 | 0.249 | 15.767 | 64.000 | 0.246 |
| 128 | 0.170 | 23.122 | 128.000 | 0.181 |
| 256 | 0.187 | 21.077 | 256.000 | 0.082 |
What is the core count where you get the most efficient use of resources?
Just using a single core is the cheapest (and always will be unless your speedup is better than perfect – “super-linear” speedup). However, it may not be possible to run on small numbers of cores depending on how much memory you need or other technical constraints. Note: on most high-end systems, nodes are not shared between users. This means you are charged for all the CPU-cores on a node regardless of whether you actually use them. Typically we would be running on many hundreds of CPU-cores not a few tens, so the real question in practice is: what is the optimal number of nodes to use? ### What is the core count where you get the fastest solution, irrespective of efficiency? 256 cores gives the fastest time to solution. The fastest time to solution does not often make the most efficient use of resources so to use this option, you may end up wasting your resources. Sometimes, when there is time pressure to run the calculations, this may be a valid approach to running applications. ### What do you think a good core count choice would be for this application to use?
8 cores is probably a good number of cores to use with a parallel efficiency of 86%. Usually, the best choice is one that delivers good parallel efficiency with an acceptable time to solution. Note that acceptable time to solution differs depending on circumstances so this is something that the individual researcher will have to assess. Good parallel efficiency is often considered to be 70% or greater though many researchers will be happy to run in a regime with parallel efficiency greater than 60%. As noted above, running with worse parallel efficiency may also be useful if the time to solution is an overriding factor.
Tips
Here are a few tips to help you use resources effectively and efficiently on HPC systems:
- Know what your priority is: do you want the results as fast as possible or are you happy to wait longer but get more research for the resources you have been allocated?
- Use your real research application to benchmark but try to shorten the run so you can turn around your benchmarking runs in a short timescale. Ideally, it should run for 10-30 minutes; short enough to run quickly but long enough so the performance is not dominated by static startup overheads (though this is application dependent). Ways to do this potentially include, for example: using a smaller number of time steps, restricting the number of SCF cycles, restricting the number of optimisation steps.
- Use basic benchmarking to help define the best resource use for your application. One useful strategy: take the core count you are using as the baseline, halve the number of cores/nodes and rerun and then double the number of cores/nodes from your baseline and rerun. Use the three data points to assess your efficiency and the impact of different core/node counts.
Key Points
- “The smaller your job, the faster it will schedule.”
Content from Using shared resources responsibly
Last updated on 2025-01-14 | Edit this page
Estimated time: 20 minutes
Overview
Questions
- How can I be a responsible user?
- How can I protect my data?
- How can I best get large amounts of data off an HPC system?
Objectives
- Learn how to be a considerate shared system citizen.
- Understand how to protect your critical data.
- Appreciate the challenges with transferring large amounts of data off HPC systems.
- Understand how to convert many files to a single archive file using tar.
One of the major differences between using remote HPC resources and your own system (e.g. your laptop) is that remote resources are shared. How many users the resource is shared between at any one time varies from system to system but it is unlikely you will ever be the only user logged into or using such a system.
The widespread usage of scheduling systems where users submit jobs on HPC resources is a natural outcome of the shared nature of these resources. There are other things you, as an upstanding member of the community, need to consider.
Be Kind to the Login Nodes
The login node is often busy managing all of the logged in users, creating and editing files and compiling software. If the machine runs out of memory or processing capacity, it will become very slow and unusable for everyone. While the machine is meant to be used, be sure to do so responsibly — in ways that will not adversely impact other users’ experience.
Login nodes are always the right place to launch jobs. Cluster policies vary, but they may also be used for proving out workflows, and in some cases, may host advanced cluster-specific debugging or development tools. The cluster may have modules that need to be loaded, possibly in a certain order, and paths or library versions that differ from your laptop, and doing an interactive test run on the head node is a quick and reliable way to discover and fix these issues.
You can always use the commands top and
ps ux to list the processes that are running on the login
node along with the amount of CPU and memory they are using. If this
check reveals that the login node is somewhat idle, you can safely use
it for your non-routine processing task. If something goes wrong — the
process takes too long, or doesn’t respond — you can use the
kill command along with the PID to terminate the
process.
Login Node Etiquette
Which of these commands would be a routine task to run on the login node?
python physics_sim.pymakecreate_directories.shmolecular_dynamics_2tar -xzf R-3.3.0.tar.gz
Building software, creating directories, and unpacking software are
common and acceptable tasks for the login node: options #2
(make), #3 (mkdir), and #5 (tar)
are probably OK. Note that script names do not always reflect their
contents: before launching #3, please
less create_directories.sh and make sure it’s not a Trojan
horse. Running resource-intensive applications is frowned upon. Unless
you are sure it will not affect other users, do not run jobs like #1
(python) or #4 (custom MD code). If you’re unsure, ask your
friendly sysadmin for advice.
If you experience performance issues with a login node you should report it to the system staff (usually via the helpdesk) for them to investigate.
Test Before Scaling
Remember that you are generally charged for usage on shared systems. A simple mistake in a job script can end up costing a large amount of resource budget. Imagine a job script with a mistake that makes it sit doing nothing for 24 hours on 1000 cores or one where you have requested 2000 cores by mistake and only use 100 of them! This problem can be compounded when people write scripts that automate job submission (for example, when running the same calculation or analysis over lots of different parameters or files). When this happens it hurts both you (as you waste lots of charged resource) and other users (who are blocked from accessing the idle compute nodes).
On very busy resources you may wait many days in a queue for your job to fail within 10 seconds of starting due to a trivial typo in the job script. This is extremely frustrating! Most systems provide dedicated resources for testing that have short wait times to help you avoid this issue.
Test Job Submission Scripts That Use Large Amounts of Resources
Before submitting a large run of jobs, submit one as a test first to make sure everything works as expected.
Before submitting a very large or very long job submit a short truncated test to ensure that the job starts as expected.
Have a Backup Plan
Although many HPC systems keep backups, it does not always cover all the file systems available and may only be for disaster recovery purposes (i.e. for restoring the whole file system if lost rather than an individual file or directory you have deleted by mistake). Protecting critical data from corruption or deletion is primarily your responsibility: keep your own backup copies.
Version control systems (such as Git) often have free, cloud-based offerings (e.g., GitHub and GitLab) that are generally used for storing source code. Even if you are not writing your own programs, these can be very useful for storing job scripts, analysis scripts and small input files.
For larger amounts of data, you should make sure you have a robust
system in place for taking copies of critical data off the HPC system
wherever possible to backed-up storage. Tools such as rsync
can be very useful for this.
Your access to the shared HPC system will generally be time-limited so you should ensure you have a plan for transferring your data off the system before your access finishes. The time required to transfer large amounts of data should not be underestimated and you should ensure you have planned for this early enough (ideally, before you even start using the system for your research).
In all these cases, the service desk of the system you are using should be able to provide useful guidance on your options for data transfer for the volumes of data you will be using.
Your Data Is Your Responsibility
Make sure you understand what the backup policy is on the file systems on the system you are using and what implications this has for your work if you lose your data on the system. Plan your backups of critical data and how you will transfer data off the system throughout the project.
On ARCHER2, the home file systems are backed up so you can restore data you deleted by mistake. A copy of the data on home file system is also kept off site for disaster recovery purposes. The work file systems are not backed up in any way.
Transferring Data
As mentioned above, many users run into the challenge of transferring large amounts of data off HPC systems at some point (this is more often in transferring data off than onto systems but the advice below applies in either case). Data transfer speed may be limited by many different factors so the best data transfer mechanism to use depends on the type of data being transferred and where the data is going.
The components between your data’s source and destination have varying levels of performance, and in particular, may have different capabilities with respect to bandwidth and latency.
Bandwidth is generally the raw amount of data per unit time a device is capable of transmitting or receiving. It’s a common and generally well-understood metric.
Latency is a bit more subtle. For data transfers, it may be thought of as the amount of time it takes to get data out of storage and into a transmittable form. Latency issues are the reason it’s advisable to execute data transfers by moving a small number of large files, rather than the converse.
Some of the key components and their associated issues are:
- Disk speed: File systems on HPC systems are often highly parallel, consisting of a very large number of high performance disk drives. This allows them to support a very high data bandwidth. Unless the remote system has a similar parallel file system you may find your transfer speed limited by disk performance at that end.
- Meta-data performance: Meta-data operations such as opening and closing files or listing the owner or size of a file are much less parallel than read/write operations. If your data consists of a very large number of small files you may find your transfer speed is limited by meta-data operations. Meta-data operations performed by other users of the system can also interact strongly with those you perform so reducing the number of such operations you use (by combining multiple files into a single file) may reduce variability in your transfer rates and increase transfer speeds.
- Network speed: Data transfer performance can be limited by network speed. More importantly it is limited by the slowest section of the network between source and destination. If you are transferring to your laptop/workstation, this is likely to be its connection (either via LAN or WiFi).
- Firewall speed: Most modern networks are protected by some form of firewall that filters out malicious traffic. This filtering has some overhead and can result in a reduction in data transfer performance. The needs of a general purpose network that hosts email/web-servers and desktop machines are quite different from a research network that needs to support high volume data transfers. If you are trying to transfer data to or from a host on a general purpose network you may find the firewall for that network will limit the transfer rate you can achieve.
As mentioned above, if you have related data that consists of a large
number of small files it is strongly recommended to pack the files into
a larger archive file for long term storage and transfer. A
single large file makes more efficient use of the file system and is
easier to move, copy and transfer because significantly fewer metadata
operations are required. Archive files can be created using tools like
tar and zip. We have already met
tar when we talked about data transfer earlier.
Consider the Best Way to Transfer Data
If you are transferring large amounts of data you will need to think about what may affect your transfer performance. It is always useful to run some tests that you can use to extrapolate how long it will take to transfer your data. Say you have a “data” folder containing 10,000 or so files, a healthy mix of small and large ASCII and binary data. Which of the following would be the best way to transfer them to ARCHER2?
- Using
scp?
- Using
rsync?
- Using
rsyncwith compression?
- Creating a
tararchive first forrsync?
BASH
[user@laptop ~]$ tar -cvf data.tar data
[user@laptop ~]$ rsync -raz data.tar userid@login.archer2.ac.uk:~/- Creating a compressed
tararchive forrsync?
Lets go through each option
-
scpwill recursively copy the directory. This works, but without compression. -
rsync -raworks likescp -r, but preserves file information like creation times. This is marginally better. -
rsync -razadds compression, which will save some bandwidth. If you have a strong CPU at both ends of the line, and you’re on a slow network, this is a good choice. - This command first uses
tarto merge everything into a single file, thenrsync -zto transfer it with compression. With this large number of files, metadata overhead can hamper your transfer, so this is a good idea. - This command uses
tar -zto compress the archive, thenrsyncto transfer it. This may perform similarly to #4, but in most cases (for large datasets), it’s the best combination of high throughput and low latency (making the most of your time and network connection).
Key Points
- “Be careful how you use the login node.”
- “Your data on the system is your responsibility.”
- “Plan and test large data transfers.”
- “It is often best to convert many files to a single archive file before transferring.”
- “Again, don’t run stuff on the login node.”