Content from Why Use HPC?
Last updated on 2025-01-14 | Edit this page
Estimated time: 30 minutes
Overview
Questions
- Why would I be interested in High Performance Computing (HPC)?
- What can I expect to learn from this course?
Objectives
- Be able to describe what an HPC system is.
- Identify how an HPC system could benefit you.
Why Use These Computers?
What do you need?
Talk to your neighbour about your research. How does computing help you do your research? How could more computing help you do more or better research?
Frequently, research problems that use computing can outgrow the desktop or laptop computer where they started:
- A statistics student wants to do cross-validate their model. This involves running the model 1000 times — but each run takes an hour. Running on their laptop will take over a month!
- A genomics researcher has been using small datasets of sequence data, but soon will be receiving a new type of sequencing data that is 10 times as large. It’s already challenging to open the datasets on their computer — analysing these larger datasets will probably crash it.
- An engineer is using a fluid dynamics package that has an option to run in parallel. So far, they haven’t used this option on their desktop, but in going from 2D to 3D simulations, simulation time has more than tripled and it might be useful to take advantage of that feature.
In all these cases, what is needed is access to more computers than can be used at the same time. Luckily, large scale computing systems — shared computing resources with lots of computers — are available at many universities, labs, or through national networks. These resources usually have more central processing units(CPUs), CPUs that operate at higher speeds, more memory, more storage, and faster connections with other computer systems. They are frequently called “clusters”, “supercomputers” or resources for “high performance computing” or HPC. In this lesson, we will usually use the terminology of HPC and HPC cluster.
Using a cluster often has the following advantages for researchers:
- Speed. With many more CPU cores, often with higher performance specs, than a typical laptop or desktop, HPC systems can offer significant speed up.
- Volume. Many HPC systems have both the processing memory (RAM) and disk storage to handle very large amounts of data. Terabytes of RAM and petabytes of storage are available for research projects.
- Efficiency. Many HPC systems operate a pool of resources that are drawn on by many users. In most cases when the pool is large and diverse enough the resources on the system are used almost constantly.
- Cost. Bulk purchasing and government funding mean that the cost to the research community for using these systems in significantly less that it would be otherwise.
- Convenience. Maybe your calculations just take a long time to run or are otherwise inconvenient to run on your personal computer. There’s no need to tie up your own computer for hours when you can use someone else’s instead.
This is how a large-scale compute system like a cluster can help solve problems like those listed at the start of the lesson.
Thinking ahead
How do you think using a large-scale computing system will be different from using your laptop? Talk to your neighbour about some differences you may already know about, and some differences/difficulties you imagine you may run into.
On Command Line
Using HPC systems often involves the use of a shell through a command line interface (CLI) and either specialized software or programming techniques. The shell is a program with the special role of having the job of running other programs rather than doing calculations or similar tasks itself. What the user types goes into the shell, which then figures out what commands to run and orders the computer to execute them. (Note that the shell is called “the shell” because it encloses the operating system in order to hide some of its complexity and make it simpler to interact with.) The most popular Unix shell is Bash, the Bourne Again SHell (so-called because it’s derived from a shell written by Stephen Bourne). Bash is the default shell on most modern implementations of Unix and in most packages that provide Unix-like tools for Windows.
Interacting with the shell is done via a command line interface (CLI) on most HPC systems. In the earliest days of computers, the only way to interact with early computers was to rewire them. From the 1950s to the 1980s most people used line printers. These devices only allowed input and output of the letters, numbers, and punctuation found on a standard keyboard, so programming languages and software interfaces had to be designed around that constraint and text-based interfaces were the way to do this. A typing-based interface is often called a command-line interface, or CLI, to distinguish it from a graphical user interface, or GUI, which most people now use. The heart of a CLI is a read-evaluate-print loop, or REPL: when the user types a command and then presses the Enter key, the computer reads it, executes it, and prints its output. The user then types another command, and so on until the user logs off.
Learning to use Bash or any other shell sometimes feels more like programming than like using a mouse. Commands are terse (often only a couple of characters long), their names are frequently cryptic, and their output is lines of text rather than something visual like a graph. However, using a command line interface can be extremely powerful, and learning how to use one will allow you to reap the benefits described above.
The rest of this lesson
The only way to use these types of resources is by learning to use the command line. This introduction to HPC systems has two parts:
- We will learn to use the UNIX command line or shell (commonly just called Bash - although this is just one implementation).
- We will use our new Bash skills to connect to and operate a high-performance computing supercomputer.
The skills we learn here have other uses beyond just HPC: Bash and UNIX skills are used everywhere, be it for web development, running software, or operating servers. It’s become so essential that Microsoft now ships it as part of Windows! Knowing how to use Bash and HPC systems will allow you to operate virtually any modern device. With all of this in mind, let’s connect to a cluster and get started!
Key Points
- High Performance Computing (HPC) typically involves connecting to very large computing systems elsewhere in the world.
- These HPC systems can be used to do work that would either be impossible or much slower or smaller systems.
- The standard method of interacting with such systems is via a command line interface such as Bash.
Content from Connecting to the remote HPC system
Last updated on 2025-01-14 | Edit this page
Estimated time: 40 minutes
Overview
Questions
- How do I open a terminal?
- How do I connect to a remote computer?
- What is an SSH key?
Objectives
- Connect to a remote HPC system.
Opening a Terminal
Connecting to an HPC system is most often done through a tool known as “SSH” (Secure SHell) and usually SSH is run through a terminal. So, to begin using an HPC system we need to begin by opening a terminal. Different operating systems have different terminals, none of which are exactly the same in terms of their features and abilities while working on the operating system. When connected to the remote system the experience between terminals will be identical as each will faithfully present the same experience of using that system.
Here is the process for opening a terminal in each operating system.
Creating a SSH key
SSH keys are an alternative method for authentication to obtain access to remote computing systems. They can also be used for authentication when transferring files or for accessing version control systems. In this section you will create a pair of SSH keys, a private key which you keep on your own computer and a public key which is placed on the remote HPC system that you will log in to.
Linux, Mac and Windows Subsystem for Linux
Once you have opened a terminal check for existing SSH keys and filenames since existing SSH keys are overwritten,
then generate a new public-private key pair,
-
-o(no default): use the OpenSSH key format, rather than PEM. -
-a(default is 16): number of rounds of passphrase derivation; increase to slow down brute force attacks. -
-t(default is rsa): specify the “type” or cryptographic algorithm. ed25519 is faster and shorter than RSA for comparable strength. -
-f(default is /home/user/.ssh/id_algorithm): filename to store your keys. If you already have SSH keys, make sure you specify a different name:ssh-keygenwill overwrite the default key if you don’t specify!
The flag -b sets the number of bits in the key. The
default is 2048. EdDSA uses a fixed key length, so this flag would have
no effect.
When prompted, enter a strong password that you will remember. Cryptography is only as good as the weakest link, and this will be used to connect to a powerful, precious, computational resource.
Take a look in ~/.ssh (use ls ~/.ssh). You
should see the two new files: your private key
(~/.ssh/key_ARCHER2_rsa) and the public key
(~/.ssh/key_ARCHER2_rsa.pub). If a key is requested by the
system administrators, the public key is the one to
provide.
PRIVATE KEYS ARE PRIVATE
A private key that is visible to anyone but you should be considered compromised, and must be destroyed. This includes having improper permissions on the directory it (or a copy) is stored in, traversing any network in the clear, attachment on unencrypted email, and even displaying the key (which is ASCII text) in your terminal window.
Protect this key as if it unlocks your front door. In many ways, it does.
Further information
For more information on SSH security and some of the flags set here, an excellent resource is Secure Secure Shell.
Windows
On Windows you can use
- puttygen, see the Putty documentation
- MobaKeyGen, see the MobaXterm documentation
Logging onto the system
With all of this in mind, let’s connect to a remote HPC system. In this workshop, we will connect to ARCHER2 — an HPC system located at the University of Edinburgh. Although it’s unlikely that every system will be exactly like ARCHER2, it’s a very good example of what you can expect from an HPC installation. To connect to our example computer, we will use SSH (if you are using PuTTY, see above).
SSH allows us to connect to UNIX computers remotely, and use them as
if they were our own. The general syntax of the connection command
follows the format
ssh -i ~/.ssh/key_for_remote_computer yourUsername@remote.computer.address
when using SSH keys and
ssh yourUsername@some.computer.address if only password
access is available. Let’s attempt to connect to the HPC system now:
or
or if SSH keys have not been enabled
OUTPUT
This node is running Cray's Linux Environment version 1.3.2
#######################################################################################
@@@@@@@@@
@@@ @@@ _ ____ ____ _ _ _____ ____ ____
@@@ @@@@@ @@@ / \ | _ \ / ___| | | | | | ____| | _ \ |___ \
@@@ @@ @@ @@@ / _ \ | |_) | | | | |_| | | _| | |_) | __) |
@@ @@ @@@ @@ @@ / ___ \ | _ < | |___ | _ | | |___ | _ < / __/
@@ @@ @@@ @@ @@ /_/ \_\ |_| \_\ \____| |_| |_| |_____| |_| \_\ |_____|
@@@ @@ @@ @@@
@@@ @@@@@ @@@ https://www.archer2.ac.uk/support-access/
@@@ @@@
@@@@@@@@@
- U K R I - E P C C - H P E C r a y -
Hostname: uan01
Distribution: SLES 15.1 1
CPUS: 256
Memory: 257.4GB
Configured: 2021-04-27
######################################################################################If you’ve connected successfully, you should see a prompt like the one below. This prompt is informative, and lets you grasp certain information at a glance. (If you don’t understand what these things are, don’t worry! We will cover things in depth as we explore the system further.)
Telling the Difference between the Local Terminal and the Remote Terminal
You may have noticed that the prompt changed when you logged into the
remote system using the terminal (if you logged in using PuTTY this will
not apply because it does not offer a local terminal). This change is
important because it makes it clear on which system the commands you
type will be run when you pass them into the terminal. This change is
also a small complication that we will need to navigate throughout the
workshop. Exactly what is reported before the $ in the
terminal when it is connected to the local system and the remote system
will typically be different for every user. We still need to indicate
which system we are entering commands on though so we will adopt the
following convention:
-
[local]$when the command is to be entered on a terminal connected to your local computer -
userid@ln03:~>when the command is to be entered on a terminal connected to the remote system -
$when it really doesn’t matter which system the terminal is connected to.
Being certain which system your terminal is connected to
If you ever need to be certain which system a terminal you are using
is connected to then use the hostname command.
Keep two terminal windows open
It is strongly recommended that you have two terminals open, one
connected to the local system and one connected to the remote system,
that you can switch back and forth between. If you only use one terminal
window then you will need to reconnect to the remote system using one of
the methods above when you see a change from [local]$ to userid@ln03:~> and
disconnect when you see the reverse.
Content from Moving around and looking at things
Last updated on 2025-01-14 | Edit this page
Estimated time: 20 minutes
Overview
Questions
- How do I navigate and look around the system?
Objectives
- Learn how to navigate around directories and look at their contents
- Explain the difference between a file and a directory.
- Translate an absolute path into a relative path and vice versa.
- Identify the actual command, flags, and filenames in a command-line call.
- Demonstrate the use of tab completion, and explain its advantages.
First commands
At this point in the lesson, we’ve just logged into the system. Nothing has happened yet, and we’re not going to be able to do anything until we learn a few basic commands. By the end of this lesson, you will know how to “move around” the system and look at what’s there.
Right now, all we see is something that looks like this:
The dollar sign is a prompt, which shows us that the shell is waiting for input; your shell may use a different character as a prompt and may add information before the prompt. When typing commands, either from these lessons or from other sources, do not type the prompt, only the commands that follow it.
Type the command whoami, then press the Enter
key (sometimes marked Return) to send the command to the shell. The
command’s output is the ID of the current user, i.e., it shows us who
the shell thinks we are:
OUTPUT
yourUsernameMore specifically, when we type whoami into the
shell:
- finds a program called
whoami, - runs that program,
- displays that program’s output, then
- displays a new prompt to tell us that it’s ready for more commands.
Next, let’s find out where we are by running a command called
pwd (which stands for “print working directory”).
(“Directory” is another word for “folder”). At any moment, our
current working directory (where we are) is the
directory that the computer assumes we want to run commands in unless we
explicitly specify something else. Here, the computer’s response is
/home/ta158/ta158/yourUsername, which is your user’s
home directory. Note that the absolute location of your
home directory may differ from system to system.
OUTPUT
/home/ta158/ta158/yourUsernameList directory contents
So, we know where we are. How do we look and see what’s in our current directory?
ls prints the names of the files and directories in the
current directory in alphabetical order, arranged neatly into
columns.
Differences between remote and local system
Open a second terminal window on your local computer and run the
ls command without logging in remotely. What differences do
you see?
You would likely see something more like this:
OUTPUT
Applications Documents Library Music Public
Desktop Downloads Movies PicturesIn addition you should also note that the preamble before the prompt
($) is different. This is very important for making sure
you know what system you are issuing commands on when in the shell.
Making directories
If nothing shows up when you run ls, it means that
nothing’s there. Let’s make a directory for us to play with.
mkdir <new directory name> makes a new directory
with that name in your current location. Notice that this command
required two pieces of input: the actual name of the command
(mkdir) and an argument that specifies the name of the
directory you wish to create.
Let’s us ls again. What do we see?
Our folder is there, awesome. What if we wanted to go inside it and
do stuff there? We will use the cd (change directory)
command to move around. Let’s cd into our new documents
folder.
OUTPUT
~/documentsThe tilde ~
What is the ~ character? When using the shell,
~ is a shortcut that represents
/home/ta158/ta158/yourUserName.
Now that we know how to use cd, we can go anywhere.
That’s a lot of responsibility. What happens if we get “lost” and want
to get back to where we started?
To go back to your home directory, the following three commands will work:
A quick note on the structure of a UNIX
(Linux/Mac/Android/Solaris/etc) filesystem. Directories and absolute
paths (i.e. exact position in the system) are always prefixed with a
/. / by itself is the “root” or base
directory.
Let’s go there now, look around, and then return to our home directory.
OUTPUT
bin dev initrd local mnt proc root scratch tmp work
boot etc lib localscratch nix project run srv usr
cvmfs home lib64 media opt ram sbin sys varThe “home” directory is the one where we generally want to keep all of our files. Other folders on a UNIX OS contain system files, and get modified and changed as you install new software or upgrade your OS.
Using HPC filesystems
On HPC systems, you have a number of places where you can store your files. These differ in both the amount of space allocated and whether or not they are backed up. File storage locations:
- Network filesystem - Your home directory is an example of a network filesystem. Data stored here is available throughout the HPC system and files stored here are often backed up (but check your local configuration to be sure!). Files stored here are typically slower to access, the data is actually stored on another computer and is being transmitted and made available over the network!
- Scratch - Some systems may offer “scratch” space. Scratch space is typically faster to use than your home directory or network filesystem, but is not usually backed up, and should not be used for long term storage.
- Work file system - As an alternative to (or sometimes as well as) Scratch space, some HPC systems offer fast file system access as a work file system. Typically, this will have higher performance than your home directory or network file system and may not be backed up. It differs from scratch space in that files in a work file system are not automatically deleted for you, you must manage the space yourself.
- Local scratch (job only) - Some systems may offer local scratch space while executing a job. (A job is a program which you submit to run on an HPC system, and will be covered later.) Such storage is very fast, but will be deleted at the end of your job.
- Ramdisk (job only) - Some systems may let you store files in a “RAM disk” while running a job, where files are stored directly in the computer’s memory. This extremely fast, but files stored here will count against your job’s memory usage and be deleted at the end of your job.
Navigation shortcuts
There are several other useful shortcuts you should be aware of.
-
.represents your current directory -
..represents the “parent” directory of your current location - While typing nearly anything, you can have bash try to autocomplete what you are typing by pressing the tab key.
Let’s try these out now:
OUTPUT
/home/ta158/ta158/yourUserName/documents
/home/ta158/ta158/yourUserNameCommand flags and options
Many commands also have multiple behaviours that you can invoke with command line ‘flags.’ What is a flag? It’s generally just your command followed by a ‘-’ and the name of the flag (sometimes it’s ‘–’ followed by the name of the flag). You follow the flag(s) with any additional arguments you might need.
We’re going to demonstrate a couple of these “flags” using
ls.
Show hidden files with -a. Hidden files are files that
begin with ., these files will not appear otherwise, but
that doesn’t mean they aren’t there! “Hidden” files are not hidden for
security purposes, they are usually just config files and other
tempfiles that the user doesn’t necessarily need to see all the
time.
OUTPUT
. .. .bash_logout .bash_profile .bashrc documents .emacs .mozilla .sshNotice how both . and .. are visible as
hidden files. Show files, their size in bytes, date last modified,
permissions, and other things with -l.
OUTPUT
drwxr-xr-x 2 yourUsername tc001 4096 Jan 14 17:31 documentsThis is a lot of information to take in at once, but we will explain
this later! ls -l is extremely useful, and tells
you almost everything you need to know about your files without actually
looking at them.
We can also use multiple flags at the same time!
OUTPUT
userid@ln03:~> ls -la
total 36
drwx--S--- 5 yourUsername tc001 4096 Nov 28 09:58 .
drwxr-x--- 3 root tc001 4096 Nov 28 09:40 ..
-rw-r--r-- 1 yourUsername tc001 18 Dec 6 2016 .bash_logout
-rw-r--r-- 1 yourUsername tc001 193 Dec 6 2016 .bash_profile
-rw-r--r-- 1 yourUsername tc001 231 Dec 6 2016 .bashrc
drwxr-sr-x 2 yourUsername tc001 4096 Nov 28 09:58 documents
-rw-r--r-- 1 yourUsername tc001 334 Mar 3 2017 .emacs
drwxr-xr-x 4 yourUsername tc001 4096 Aug 2 2016 .mozilla
drwx--S--- 2 yourUsername tc001 4096 Nov 28 09:58 .sshFlags generally precede any arguments passed to a UNIX command.
ls actually takes an extra argument that specifies a
directory to look into. When you use flags and arguments together, the
syntax (how it’s supposed to be typed) generally looks something like
this:
So using ls -l -a on a different directory than the one
we’re in would look something like:
OUTPUT
drwxr-sr-x 2 yourUsername tc001 4096 Nov 28 09:58 .
drwx--S--- 5 yourUsername tc001 4096 Nov 28 09:58 ..Where to go for help?
How did I know about the -l and -a options?
Is there a manual we can look at for help when we need help? There is a
very helpful manual for most UNIX commands: man (if you’ve
ever heard of a “man page” for something, this is what it is).
OUTPUT
LS(1) User Commands LS(1)
NAME
ls - list directory contents
SYNOPSIS
ls [OPTION]... [FILE]...
DESCRIPTION
List information about the FILEs (the current directory by default).
Sort entries alphabetically if none of -cftuvSUX nor --sort is specified.
Mandatory arguments to long options are mandatory for short options too.To navigate through the man pages, you may use the up
and down arrow keys to move line-by-line, or try the Space
and b keys to skip up and down by full page. Quit the
man pages by typing q.
Alternatively, most commands you run will have a --help
option that displays addition information For instance, with
ls:
OUTPUT
Usage: ls [OPTION]... [FILE]...
List information about the FILEs (the current directory by default).
Sort entries alphabetically if none of -cftuvSUX nor --sort is specified.
Mandatory arguments to long options are mandatory for short options too.
-a, --all do not ignore entries starting with .
-A, --almost-all do not list implied . and ..
--author with -l, print the author of each file
-b, --escape print C-style escapes for nongraphic characters
--block-size=SIZE scale sizes by SIZE before printing them; e.g.,
'--block-size=M' prints sizes in units of
1,048,576 bytes; see SIZE format below
-B, --ignore-backups do not list implied entries ending with ~
# further output omitted for clarityUnsupported command-line options
If you try to use an option that is not supported, ls
and other programs will print an error message similar to this:
ERROR
ls: invalid option -- 'j'
Try 'ls --help' for more information.- No:
.stands for the current directory. - No:
/stands for the root directory. - No: Amanda’s home directory is
/Users/amanda. - No: this goes up two levels, i.e. ends in
/Users. - Yes:
~stands for the user’s home directory, in this case/Users/amanda. - No: this would navigate into a directory
homein the current directory if it exists. - Yes: unnecessarily complicated, but correct.
- Yes: shortcut to go back to the user’s home directory.
- Yes: goes up one level.
Relative Path Resolution
Using the filesystem diagram below, if pwd displays
/Users/thing, what will ls -F ../backup
display?
../backup: No such file or directory2012-12-01 2013-01-08 2013-01-272012-12-01/ 2013-01-08/ 2013-01-27/original/ pnas_final/ pnas_sub/

- No: there is a directory
backupin/Users. - No: this is the content of
Users/thing/backup, but with..we asked for one level further up. - No: see previous explanation.
- Yes:
../backup/refers to/Users/backup/.
ls Reading Comprehension
Assuming a directory structure as in the above Figure (File System
for Challenge Questions), if pwd displays
/Users/backup, and -r tells ls to
display things in reverse order, what command will display:
OUTPUT
pnas_sub/ pnas_final/ original/ls pwdls -r -Fls -r -F /Users/backup- Either #2 or #3 above, but not #1.
- No:
pwdis not the name of a directory. - Yes:
lswithout directory argument lists files and directories in the current directory. - Yes: uses the absolute path explicitly.
- Correct: see explanations above.
Exploring More ls Arguments
What does the command ls do when used with the
-l and -h arguments? Some of its output is
about properties that we do not cover in this lesson (such as file
permissions and ownership), but the rest should be useful
nevertheless.
The -l arguments makes ls use a
long listing format, showing not only the
file/directory names but also additional information such as the file
size and the time of its last modification. The -h argument
makes the file size “human readable”, i.e. display
something like 5.3K instead of 5369.
Listing Recursively and By Time
The command ls -R lists the contents of directories
recursively, i.e., lists their sub-directories, sub-sub-directories, and
so on in alphabetical order at each level. The command
ls -t lists things by time of last change, with most
recently changed files or directories first. In what order does
ls -R -t display things? Hint: ls -l uses a
long listing format to view timestamps.
The directories are listed alphabetical at each level, the files/directories in each directory are sorted by time of last change.
Key Points
- Your current directory is referred to as the working directory.
- To change directories, use
cd. - To view files, use
ls. - You can view help for a command with
man commandorcommand --help. - Hit Tab to autocomplete whatever you’re currently typing.
Content from Working with files
Last updated on 2025-01-14 | Edit this page
Estimated time: 45 minutes
Overview
Questions
- How do I create/edit text files?
- How do I move/copy/delete files?
Objectives
- Learn to use the
nanotext editor. - Understand how to move, create, and delete files.
Now that we know how to move around and look at things, let’s learn how to read, write, and handle files! We’ll start by moving back to our home directory and creating a scratch directory:
Creating and Editing Text Files
When working on an HPC system, we will frequently need to create or edit text files. Text is one of the simplest computer file formats, defined as a simple sequence of text lines.
What if we want to make a file? There are a few ways of doing this,
the easiest of which is simply using a text editor. For this lesson, we
are going to us nano, since it’s more intuitive than many
other terminal text editors.
To create or edit a file, type nano <filename>, on
the terminal, where <filename> is the name of the
file. If the file does not already exist, it will be created. Let’s make
a new file now, type whatever you want in it, and save it.
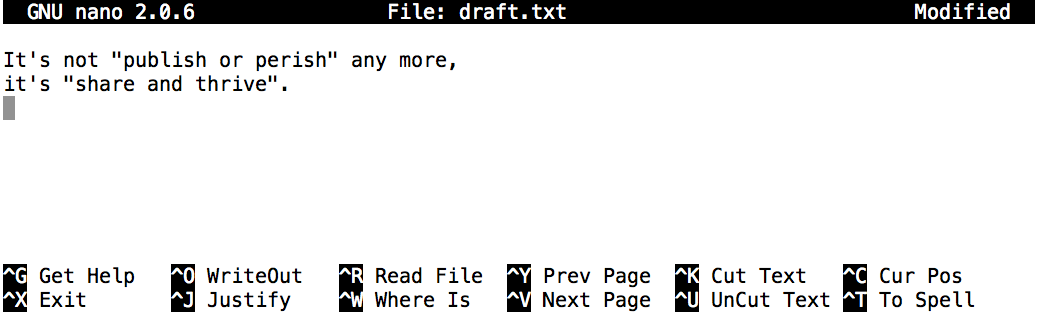
Nano defines a number of shortcut keys (prefixed by the Control or Ctrl key) to perform actions such as saving the file or exiting the editor. Here are the shortcut keys for a few common actions:
Ctrl+O — save the file (into a current name or a new name).
Ctrl+X — exit the editor. If you have not saved your file upon exiting,
nanowill ask you if you want to save.Ctrl+K — cut (“kill”) a text line. This command deletes a line and saves it on a clipboard. If repeated multiple times without any interruption (key typing or cursor movement), it will cut a chunk of text lines.
Ctrl+U — paste the cut text line (or lines). This command can be repeated to paste the same text elsewhere.
Using vim as a text editor
From time to time, you may encounter the vim text
editor. Although vim isn’t the easiest or most
user-friendly of text editors, you’ll be able to find it on any system
and it has many more features than nano.
vim has several modes, a “command” mode (for doing big
operations, like saving and quitting) and an “insert” mode. You can
switch to insert mode with the i key, and command mode with
Esc.
In insert mode, you can type more or less normally. In command mode there are a few commands you should be aware of:
-
:q!— quit, without saving -
:wq— save and quit -
dd— cut/delete a line -
y— paste a line
Do a quick check to confirm our file was created.
OUTPUT
draft.txtReading Files
Let’s read the file we just created now. There are a few different
ways of doing this, one of which is reading the entire file with
cat.
OUTPUT
It's not "publish or perish" any more,
it's "share and thrive".By default, cat prints out the content of the given
file. Although cat may not seem like an intuitive command
with which to read files, it stands for “concatenate”. Giving it
multiple file names will print out the contents of the input files in
the order specified in the cat’s invocation. For
example,
OUTPUT
It's not "publish or perish" any more,
it's "share and thrive".
It's not "publish or perish" any more,
it's "share and thrive".Reading Multiple Text Files
Create two more files using nano, giving them different
names such as chap1.txt and chap2.txt. Then
use a single cat command to read and print the contents of
draft.txt, chap1.txt, and
chap2.txt.
Creating Directory
We’ve successfully created a file. What about a directory? We’ve
actually done this before, using mkdir.
OUTPUT
draft.txt filesMoving, Renaming, Copying Files
Moving — We will move draft.txt to the
files directory with mv (“move”) command. The
same syntax works for both files and directories:
mv <file/directory> <new-location>
OUTPUT
draft.txtRenaming — draft.txt isn’t a very
descriptive name. How do we go about changing it? It turns out that
mv is also used to rename files and directories. Although
this may not seem intuitive at first, think of it as moving a
file to be stored under a different name. The syntax is quite similar to
moving files: mv oldName newName.
OUTPUT
newname.testfileRemoving files
We’ve begun to clutter up our workspace with all of the directories and files we’ve been making. Let’s learn how to get rid of them. One important note before we start… when you delete a file on UNIX systems, they are gone forever. There is no “recycle bin” or “trash”. Once a file is deleted, it is gone, never to return. So be very careful when deleting files.
Files are deleted with rm file [moreFiles]. To delete
the newname.testfile in our current directory:
OUTPUT
files Documents newname.testfile
files DocumentsThat was simple enough. Directories are deleted in a similar manner
using rm -r (the -r option stands for
‘recursive’).
OUTPUT
files Documents
rmdir: failed to remove `files/': Directory not empty
filesWhat happened? As it turns out, rmdir is unable to
remove directories that have stuff in them. To delete a directory and
everything inside it, we will use a special variant of rm,
rm -rf directory. This is probably the scariest command on
UNIX- it will force delete a directory and all of its contents without
prompting. ALWAYS double check your typing before using
it… if you leave out the arguments, it will attempt to delete everything
on your file system that you have permission to delete. So when deleting
directories be very, very careful.
What happens when you use rm -rf
accidentally
Steam is a major online sales platform for PC video games with over 125 million users. Despite this, it hasn’t always had the most stable or error-free code.
In January 2015, user kevyin on GitHub reported
that Steam’s Linux client had deleted every file on his computer. It
turned out that one of the Steam programmers had added the following
line: rm -rf "$STEAMROOT/"*. Due to the way that Steam was
set up, the variable $STEAMROOT was never initialized,
meaning the statement evaluated to rm -rf /*. This coding
error in the Linux client meant that Steam deleted every single file on
a computer when run in certain scenarios (including connected external
hard drives). Moral of the story: be very careful when
using rm -rf!
Looking at files
Sometimes it’s not practical to read an entire file with
cat- the file might be way too large, take a long time to
open, or maybe we want to only look at a certain part of the file. As an
example, we are going to look at a large and complex file type used in
bioinformatics- a .gtf file. The GTF2 format is commonly
used to describe the location of genetic features in a genome.
Let’s grab and unpack a set of demo files for use later. To do this,
we’ll use wget
(wget <link> downloads a file from a link).
Problems with wget?
wget is a stand-alone application for downloading things
over HTTP/HTTPS and FTP/FTPS connections, and it does the job admirably
— when it is installed.
Some operating systems instead come with cURL, which is the command-line
interface to libcurl, a powerful library for programming
interactions with remote resources over a wide variety of network
protocols. If you have curl but not wget, then
try this command instead:
For very large downloads, you might consider using Aria2, which has support for downloading the same file from multiple mirrors. You have to install it separately, but if you have it, try this to get it faster than your neighbors:
macOS:
curlis pre-installed on macOS. If you must have the latest version you canbrew installit, but only do so if the stock version has failed you.Windows:
curlcomes preinstalled for the Windows 10 command line. For earlier Windows systems, you can download the executable directly; run it in place.curlcomes preinstalled in Git for Windows and Windows Subsystem for Linux. On Cygwin, run the setup program again and select thecurlpackage to install it.-
Linux:
curlis packaged for every major distribution. You can install it through the usual means.- Debian, Ubuntu, Mint:
sudo apt install curl - CentOS, Red Hat:
sudo yum install curlorzypper install curl - Fedora:
sudo dnf install curl
- Debian, Ubuntu, Mint:
macOS:
aria2cis available through a homebrew.brew install aria2.Windows: download the latest release and run
aria2cin place. If you’re using the Windows Subsystem for Linux,-
Linux: every major distribution has an
aria2package. Install it by the usual means.- Debian, Ubuntu, Mint:
sudo apt install aria2 - CentOS, Red Hat:
sudo yum install aria2orzypper install aria2 - Fedora:
sudo dnf install aria2
- Debian, Ubuntu, Mint:
You’ll commonly encounter .tar.gz archives while working
in UNIX. To extract the files from a .tar.gz file, we run
the command tar -xvf filename.tar.gz:
OUTPUT
dmel-all-r6.19.gtf
dmel_unique_protein_isoforms_fb_2016_01.tsv
gene_association.fb
SRR307023_1.fastq
SRR307023_2.fastq
SRR307024_1.fastq
SRR307024_2.fastq
SRR307025_1.fastq
SRR307025_2.fastq
SRR307026_1.fastq
SRR307026_2.fastq
SRR307027_1.fastq
SRR307027_2.fastq
SRR307028_1.fastq
SRR307028_2.fastq
SRR307029_1.fastq
SRR307029_2.fastq
SRR307030_1.fastq
SRR307030_2.fastqUnzipping files
We just unzipped a .tar.gz file for this example. What if we run into other file formats that we need to unzip? Just use the handy reference below:
-
gunzipextracts the contents of .gz files -
unzipextracts the contents of .zip files -
tar -xvfextracts the contents of .tar.gz and .tar.bz2 files
That is a lot of files! One of these files,
dmel-all-r6.19.gtf is extremely large, and contains every
annotated feature in the Drosophila melanogaster genome. It’s a
huge file- what happens if we run cat on it? (Press
Ctrl + c to stop it).
So, cat is a really bad option when reading big files…
it scrolls through the entire file far too quickly! What are the
alternatives? Try all of these out and see which ones you like best!
-
head <file>: Print the top 10 lines in a file to the console. You can control the number of lines you see with the-n <numberOfLines>flag. -
tail <file>: Same ashead, but prints the last 10 lines in a file to the console. -
less <file>: Opens a file and display as much as possible on-screen. You can scroll with Enter or the arrow keys on your keyboard. Press q to close the viewer.
Challenge
Out of cat, head, tail, and
less, which method of reading files is your favourite?
Why?
Key Points
- Use
nanoto create or edit text files from a terminal. - Use
cat file1 [file2 ...]to print the contents of one or more files to the terminal. - Use
mv old dirto move a file or directoryoldto another directorydir. - Use
mv old newto rename a file or directoryoldto anewname. - Use
cp old newto copy a file under a new name or location. - Use
cp old dircopies a fileoldinto a directorydir. - Use
rm oldto delete (remove) a file. - File extensions are entirely arbitrary on UNIX systems.
Content from Wildcards and pipes
Last updated on 2025-01-14 | Edit this page
Estimated time: 50 minutes
Overview
Questions
- How can I run a command on multiple files at once?
- Is there an easy way of saving a command’s output?
Objectives
- Redirect a command’s output to a file.
- Process a file instead of keyboard input using redirection.
- Construct command pipelines with two or more stages.
- Explain what usually happens if a program or pipeline isn’t given any input to process.
Required files
If you didn’t get them in the last lesson, make sure to download the example files used in the next few sections:
Using wget:
Using a web browser: https://epcced.github.io/2024-06-19-hpc-shell-shampton/files/bash-lesson.tar.gz
Now that we know some of the basic UNIX commands, we are going to
explore some more advanced features. The first of these features is the
wildcard *. In our examples before, we’ve done things to
files one at a time and otherwise had to specify things explicitly. The
* character lets us speed things up and do things across
multiple files.
Ever wanted to move, delete, or just do “something” to all files of a
certain type in a directory? * lets you do that, by taking
the place of one or more characters in a piece of text. So
*.txt would be equivalent to all .txt files in
a directory for instance. * by itself means all files.
Let’s use our example data to see what I mean.
OUTPUT
bash-lesson.tar.gz SRR307026_1.fastq
dmel-all-r6.19.gtf SRR307026_2.fastq
dmel_unique_protein_isoforms_fb_2016_01.tsv SRR307027_1.fastq
gene_association.fb SRR307027_2.fastq
SRR307023_1.fastq SRR307028_1.fastq
SRR307023_2.fastq SRR307028_2.fastq
SRR307024_1.fastq SRR307029_1.fastq
SRR307024_2.fastq SRR307029_2.fastq
SRR307025_1.fastq SRR307030_1.fastq
SRR307025_2.fastq SRR307030_2.fastqWord counts and wildcards (globbing)
Now we have a whole bunch of example files in our directory. For this
example we are going to learn a new command that tells us how long a
file is: wc. wc -l file tells us the length of
a file in lines.
OUTPUT
542048 dmel-all-r6.19.gtfInteresting, there are over 540000 lines in our
dmel-all-r6.19.gtf file. What if we wanted to run
wc -l on every .fastq file? This is where
* comes in really handy! *.fastq would match
every file ending in .fastq.
OUTPUT
20000 SRR307023_1.fastq
20000 SRR307023_2.fastq
20000 SRR307024_1.fastq
20000 SRR307024_2.fastq
20000 SRR307025_1.fastq
20000 SRR307025_2.fastq
20000 SRR307026_1.fastq
20000 SRR307026_2.fastq
20000 SRR307027_1.fastq
20000 SRR307027_2.fastq
20000 SRR307028_1.fastq
20000 SRR307028_2.fastq
20000 SRR307029_1.fastq
20000 SRR307029_2.fastq
20000 SRR307030_1.fastq
20000 SRR307030_2.fastq
320000 totalThat was easy. What if we wanted to do the same command, except on
every file in the directory? A nice trick to keep in mind is that
* by itself matches every file.
OUTPUT
53037 bash-lesson.tar.gz
542048 dmel-all-r6.19.gtf
22129 dmel_unique_protein_isoforms_fb_2016_01.tsv
106290 gene_association.fb
20000 SRR307023_1.fastq
20000 SRR307023_2.fastq
20000 SRR307024_1.fastq
20000 SRR307024_2.fastq
20000 SRR307025_1.fastq
20000 SRR307025_2.fastq
20000 SRR307026_1.fastq
20000 SRR307026_2.fastq
20000 SRR307027_1.fastq
20000 SRR307027_2.fastq
20000 SRR307028_1.fastq
20000 SRR307028_2.fastq
20000 SRR307029_1.fastq
20000 SRR307029_2.fastq
20000 SRR307030_1.fastq
20000 SRR307030_2.fastq
1043504 totalMultiple wildcards
You can even use multiple *s at a time. How would you
run wc -l on every file with “fb” in it?
Using other commands
Now let’s try cleaning up our working directory a bit. Create a
folder called “fastq” and move all of our .fastq files there in one
mv command.
Redirecting output
Each of the commands we’ve used so far does only a very small amount of work. However, we can chain these small UNIX commands together to perform otherwise complicated actions!
For our first foray into piping, or redirecting output, we
are going to use the > operator to write output to a
file. When using >, whatever is on the left of the
> is written to the filename you specify on the right of
the arrow. The actual syntax looks like
command > filename.
Let’s try several basic usages of >.
echo simply prints back, or echoes whatever you type after
it.
OUTPUT
this is a test
bash-lesson.tar.gz fastq
dmel-all-r6.19.gtf gene_association.fb
dmel_unique_protein_isoforms_fb_2016_01.tsv test.txt
this is a testAwesome, let’s try that with a more complicated command, like
wc -l.
OUTPUT
wc: fastq: Is a directory
53037 bash-lesson.tar.gz
542048 dmel-all-r6.19.gtf
22129 dmel_unique_protein_isoforms_fb_2016_01.tsv
0 fastq
106290 gene_association.fb
1 test.txt
723505 totalNotice how we still got some output to the console even though we “piped” the output to a file? Our expected output still went to the file, but how did the error message get skipped and not go to the file?
This phenomena is an artefact of how UNIX systems are built. There
are 3 input/output streams for every UNIX program you will run:
stdin, stdout, and stderr.
Let’s dissect these three streams of input/output in the command we
just ran: wc -l * > word_counts.txt
-
stdinis the input to a program. In the command we just ran,stdinis represented by*, which is simply every filename in our current directory. -
stdoutcontains the actual, expected output. In this case,>redirectedstdoutto the fileword_counts.txt. -
stderrtypically contains error messages and other information that doesn’t quite fit into the category of “output”. If we insist on redirecting bothstdoutandstderrto the same file we would use&>instead of>. (We can redirect juststderrusing2>.)
Knowing what we know now, let’s try re-running the command, and send
all of the output (including the error message) to the same
word_counts.txt files as before.
Notice how there was no output to the console that time. Let’s check that the error message went to the file like we specified.
OUTPUT
53037 bash-lesson.tar.gz
542048 dmel-all-r6.19.gtf
22129 dmel_unique_protein_isoforms_fb_2016_01.tsv
wc: fastq: Is a directory
0 fastq
106290 gene_association.fb
1 test.txt
7 word_counts.txt
723512 totalSuccess! The wc: fastq: Is a directory error message was
written to the file. Also, note how the file was silently overwritten by
directing output to the same place as before. Sometimes this is not the
behaviour we want. How do we append (add) to a file instead of
overwriting it?
Appending to a file is done the same was as redirecting output.
However, instead of >, we will use
>>.
BASH
$ echo "We want to add this sentence to the end of our file" >> word_counts.txt
$ cat word_counts.txtOUTPUT
53037 bash-lesson.tar.gz
542048 dmel-all-r6.19.gtf
22129 dmel_unique_protein_isoforms_fb_2016_01.tsv
wc: fastq: Is a directory
0 fastq
106290 gene_association.fb
1 test.txt
7 word_counts.txt
723512 total
We want to add this sentence to the end of our fileChaining commands together
We now know how to redirect stdout and
stderr to files. We can actually take this a step further
and redirect output (stdout) from one command to serve as
the input (stdin) for the next. To do this, we use the
| (pipe) operator.
grep is an extremely useful command. It finds things for
us within files. Basic usage (there are a lot of options for more clever
things, see the man page) uses the syntax
grep whatToFind fileToSearch. Let’s use grep
to find all of the entries pertaining to the Act5C gene in
Drosophila melanogaster.
The output is nearly unintelligible since there is so much of it.
Let’s send the output of that grep command to
head so we can just take a peek at the first line. The
| operator lets us send output from one command to the
next:
OUTPUT
X FlyBase gene 5900861 5905399 . + . gene_id "FBgn0000042"; gene_symbol "Act5C";Nice work, we sent the output of grep to
head. Let’s try counting the number of entries for Act5C
with wc -l. We can do the same trick to send
grep’s output to wc -l:
OUTPUT
46Note that this is just the same as redirecting output to a file, then reading the number of lines from that file.
Writing commands using pipes
How many files are there in the “fastq” directory we made earlier? (Use the shell to do this.)
Reading from compressed files
Let’s compress one of our files using gzip.
zcat acts like cat, except that it can read
information from .gz (compressed) files. Using
zcat, can you write a command to take a look at the top few
lines of the gene_association.fb.gz file (without
decompressing the file itself)?
Key Points
- The
*wildcard is used as a placeholder to match any text that follows a pattern. - Redirect a command’s output to a file with
>. - Commands can be chained with
|
Content from Scripts, variables, and loops
Last updated on 2025-01-14 | Edit this page
Estimated time: 55 minutes
Overview
Questions
- How do I turn a set of commands into a program?
Objectives
- Write a shell script
- Understand and manipulate UNIX permissions
- Understand shell variables and how to use them
- Write a simple “for” loop.
We now know a lot of UNIX commands! Wouldn’t it be great if we could save certain commands so that we could run them later or not have to type them out again? As it turns out, this is straightforward to do. A “shell script” is essentially a text file containing a list of UNIX commands to be executed in a sequential manner. These shell scripts can be run whenever we want, and are a great way to automate our work.
Writing a Script
So how do we write a shell script, exactly? It turns out we can do
this with a text editor. Start editing a file called “demo.sh” (to
recap, we can do this with nano demo.sh). The “.sh” is the
standard file extension for shell scripts that most people use (you may
also see “.bash” used).
Our shell script will have two parts:
- On the very first line, add
#!/bin/bash. The#!(pronounced “hash-bang”) tells our computer what program to run our script with. In this case, we are telling it to run our script with our command-line shell (what we’ve been doing everything in so far). If we wanted our script to be run with something else, like Perl, we could add#!/usr/bin/perl - Now, anywhere below the first line, add
echo "Our script worked!". When our script runs,echowill happily print outOur script worked!.
Our file should now look like this:
Ready to run our program? Let’s try running it:
ERROR
bash: demo.sh: command not found...Strangely enough, Bash can’t find our script. As it turns out, Bash
will only look in certain directories for scripts to run. To run
anything else, we need to tell Bash exactly where to look. To run a
script that we wrote ourselves, we need to specify the full path to the
file, followed by the filename. We could do this one of two ways: either
with our absolute path
/home/ta158/ta158/yourUserName/demo.sh, or with the
relative path ./demo.sh.
ERROR
bash: ./demo.sh: Permission deniedThere’s one last thing we need to do. Before a file can be run, it
needs “permission” to run. Let’s look at our file’s permissions with
ls -l:
OUTPUT
-rw-rw-r-- 1 yourUsername tc001 12534006 Jan 16 18:50 bash-lesson.tar.gz
-rw-rw-r-- 1 yourUsername tc001 40 Jan 16 19:41 demo.sh
-rw-rw-r-- 1 yourUsername tc001 77426528 Jan 16 18:50 dmel-all-r6.19.gtf
-rw-r--r-- 1 yourUsername tc001 721242 Jan 25 2016 dmel_unique_protein_is...
drwxrwxr-x 2 yourUsername tc001 4096 Jan 16 19:16 fastq
-rw-r--r-- 1 yourUsername tc001 1830516 Jan 25 2016 gene_association.fb.gz
-rw-rw-r-- 1 yourUsername tc001 15 Jan 16 19:17 test.txt
-rw-rw-r-- 1 yourUsername tc001 245 Jan 16 19:24 word_counts.txtThat’s a huge amount of output: a full listing of everything in the directory. Let’s see if we can understand what each field of a given row represents, working left to right.
-
Permissions: On the very left side, there is a
string of the characters
d,r,w,x, and-. Thedindicates if something is a directory (there is a-in that spot if it is not a directory). The otherr,w,xbits indicate permission to Read, Write, and eXecute a file. There are three fields ofrwxpermissions following the spot ford. If a user is missing a permission to do something, it’s indicated by a-.- The first set of
rwxare the permissions that the owner has (in this case the owner isyourUsername). - The second set of
rwxs are permissions that other members of the owner’s group share (in this case, the group is namedtc001). - The third set of
rwxs are permissions that anyone else with access to this computer can do with a file. Though files are typically created with read permissions for everyone, typically the permissions on your home directory prevent others from being able to access the file in the first place.
- The first set of
- References: This counts the number of references (hard links) to the item (file, folder, symbolic link or “shortcut”).
- Owner: This is the username of the user who owns the file. Their permissions are indicated in the first permissions field.
- Group: This is the user group of the user who owns the file. Members of this user group have permissions indicated in the second permissions field.
-
Size of item: This is the number of bytes in a
file, or the number of filesystem
blocks occupied by the contents of a folder. (We can use the
-hoption here to get a human-readable file size in megabytes, gigabytes, etc.) - Time last modified: This is the last time the file was modified.
- Filename: This is the filename.
So how do we change permissions? As I mentioned earlier, we need
permission to execute our script. Changing permissions is done with
chmod. To add executable permissions for all users we could
use this:
OUTPUT
-rw-rw-r-- 1 yourUsername tc001 12534006 Jan 16 18:50 bash-lesson.tar.gz
-rwxrwxr-x 1 yourUsername tc001 40 Jan 16 19:41 demo.sh
-rw-rw-r-- 1 yourUsername tc001 77426528 Jan 16 18:50 dmel-all-r6.19.gtf
-rw-r--r-- 1 yourUsername tc001 721242 Jan 25 2016 dmel_unique_protein_is...
drwxrwxr-x 2 yourUsername tc001 4096 Jan 16 19:16 fastq
-rw-r--r-- 1 yourUsername tc001 1830516 Jan 25 2016 gene_association.fb.gz
-rw-rw-r-- 1 yourUsername tc001 15 Jan 16 19:17 test.txt
-rw-rw-r-- 1 yourUsername tc001 245 Jan 16 19:24 word_counts.txtNow that we have executable permissions for that file, we can run it.
OUTPUT
Our script worked!Fantastic, we’ve written our first program! Before we go any further,
let’s learn how to take notes inside our program using comments. A
comment is indicated by the # character, followed by
whatever we want. Comments do not get run. Let’s try out some comments
in the console, then add one to our script!
OUTPUT
# This won't show anything.Now lets try adding this to our script with nano. Edit
your script to look something like this:
When we run our script, the output should be unchanged from before!
Shell variables
One important concept that we’ll need to cover are shell variables. Variables are a great way of saving information under a name you can access later. In programming languages like Python and R, variables can store pretty much anything you can think of. In the shell, they usually just store text. The best way to understand how they work is to see them in action.
To set a variable, simply type in a name containing only letters,
numbers, and underscores, followed by an = and whatever you
want to put in the variable. Shell variable names are often uppercase by
convention (but do not have to be).
To use a variable, prefix its name with a $ sign. Note
that if we want to simply check what a variable is, we should use echo
(or else the shell will try to run the contents of a variable).
OUTPUT
This is our variableLet’s try setting a variable in our script and then recalling its
value as part of a command. We’re going to make it so our script runs
wc -l on whichever file we specify with
FILE.
Our script:
BASH
#!/bin/bash
# set our variable to the name of our GTF file
FILE=dmel-all-r6.19.gtf
# call wc -l on our file
wc -l $FILEOUTPUT
542048 dmel-all-r6.19.gtfWhat if we wanted to do our little wc -l script on other
files without having to change $FILE every time we want to
use it? There is actually a special shell variable we can use in scripts
that allows us to use arguments in our scripts (arguments are extra
information that we can pass to our script, like the -l in
wc -l).
To use the first argument to a script, use $1 (the
second argument is $2, and so on). Let’s change our script
to run wc -l on $1 instead of
$FILE. Note that we can also pass all of the arguments
using $@ (not going to use it in this lesson, but it’s
something to be aware of).
Our script:
OUTPUT
22129 dmel_unique_protein_isoforms_fb_2016_01.tsvNice! One thing to be aware of when using variables: they are all
treated as pure text. How do we save the output of an actual command
like ls -l?
A demonstration of what doesn’t work:
ERROR
-bash: -l: command not foundWhat does work (we need to surround any command with
$(command)):
OUTPUT
total 90372 -rw-rw-r-- 1 jeff jeff 12534006 Jan 16 18:50 bash-lesson.tar.gz -rwxrwxr-x. 1 jeff jeff 40 Jan 1619:41 demo.sh -rw-rw-r-- 1 jeff jeff 77426528 Jan 16 18:50 dmel-all-r6.19.gtf -rw-r--r-- 1 jeff jeff 721242 Jan 25 2016 dmel_unique_protein_isoforms_fb_2016_01.tsv drwxrwxr-x. 2 jeff jeff 4096 Jan 16 19:16 fastq -rw-r--r-- 1 jeff jeff 1830516 Jan 25 2016 gene_association.fb.gz -rw-rw-r-- 1 jeff jeff 15 Jan 16 19:17 test.txt -rw-rw-r-- 1 jeff jeff 245 Jan 16 19:24 word_counts.txtNote that everything got printed on the same line. This is a feature,
not a bug, as it allows us to use $(commands) inside lines
of script without triggering line breaks (which would end our line of
code and execute it prematurely).
Loops
To end our lesson on scripts, we are going to learn how to write a for-loop to execute a lot of commands at once. This will let us do the same string of commands on every file in a directory (or other stuff of that nature).
for-loops generally have the following syntax:
When a for-loop gets run, the loop will run once for everything
following the word in. In each iteration, the variable
$VAR is set to a particular value for that iteration. In
this case it will be set to first during the first
iteration, second on the second, and so on. During each
iteration, the code between do and done is
performed.
Let’s run the script we just wrote (I saved mine as
loop.sh).
OUTPUT
first
second
thirdWhat if we wanted to loop over a shell variable, such as every file
in the current directory? Shell variables work perfectly in for-loops.
In this example, we’ll save the result of ls and loop over
each file:
OUTPUT
bash-lesson.tar.gz
demo.sh
dmel_unique_protein_isoforms_fb_2016_01.tsv
dmel-all-r6.19.gtf
fastq
gene_association.fb.gz
loop.sh
test.txt
word_counts.txtThere’s a shortcut to run on all files of a particular type, say all
.gz files:
OUTPUT
bash-lesson.tar.gz
gene_association.fb.gzWriting our own scripts and loops
cd to our fastq directory from earlier and
write a loop to print off the name and top 4 lines of every fastq file
in that directory.
Is there a way to only run the loop on fastq files ending in
_1.fastq?
Concatenating variables
Concatenating (i.e. mashing together) variables is quite easy to do.
Add whatever you want to concatenate to the beginning or end of the
shell variable after enclosing it in {} characters.
OUTPUT
stuff.txt.exampleCan you write a script that prints off the name of every file in a directory with “.processed” added to it?
Create the following script in a file called
process.sh
Note that this will also print directories appended with
“.processed”. To truly only get files and not directories, we need to
modify this to use the find command to give us only files
in the current directory:
but this will have the side-effect of listing hidden files too.
Special permissions
What if we want to give different sets of users different
permissions. chmod actually accepts special numeric codes
instead of stuff like chmod +x. The numeric codes are as
follows: read = 4, write = 2, execute = 1. For each user we will assign
permissions based on the sum of these permissions (must be between 7 and
0).
Let’s make an example file and give everyone permission to do everything with it.
How might we give ourselves permission to do everything with a file, but allow no one else to do anything with it.
Key Points
- A shell script is just a list of bash commands in a text file.
- To make a shell script file executable, run
chmod +x script.sh.